MMSEQ: Transcript and gene level expression analysis using multi-mapping RNA-seq reads

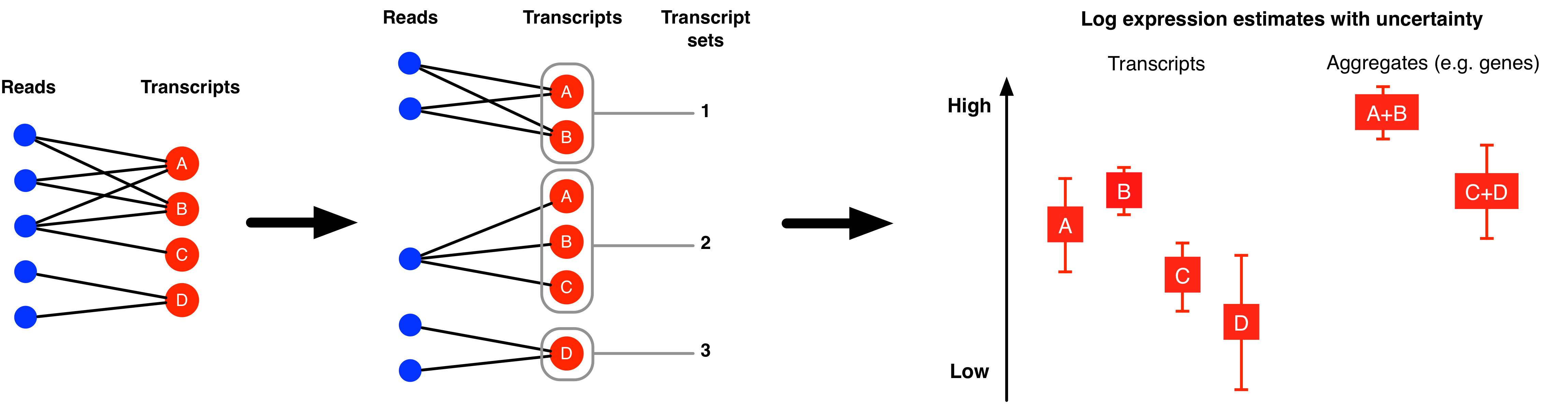
What is MMSEQ?
The MMSEQ package contains a collection of statistical tools for analysing RNA-seq expression data. Expression levels are inferred for each transcript using the mmseq program by modelling mappings of reads or read pairs (fragments) to sets of transcripts. These transcripts can be based on reference, custom or haplotype-specific sequences. The latter allows haplotype-specific analysis, which is useful in studies of allelic imbalance. The posterior distributions of the expression parameters for groups of transcripts belonging to the same gene are aggregated to provide gene-level expression estimates. Other aggregations (e.g. of transcripts sharing the same UTRs) are also possible. Isoform usage (i.e., the proportion of a gene's expression due to each isoform) is also estimated. Uncertainty in expression levels is summarised as the standard deviation of the posterior distribution of each expression parameter. When the uncertainty is large in all samples, a collapsing algorithm can be used for grouping transcripts into inferential units with reduced levels of uncertainty.
The package also includes a model-selection algorithm for differential analysis (implemented in mmdiff) that takes into account the posterior uncertainty in the expression parameters and can be used to select amongst an arbitrary number of models. The algorithm is regression-based and thus it can accomodate complex experimental designs. The model selection algorithm can be applied at the level of transcripts or transcript aggregates such as genes and it can also be applied to detect differential isoform usage by modelling summaries of the posterior distributions of isoform usage proportions as the outcomes of the linear regression models.
Citing MMSEQ
If you use the MMSEQ package, please cite:
- Haplotype and isoform specific expression estimation using multi-mapping RNA-seq reads. Turro E, Su S-Y, Goncalves A, Coin L, Richardson S and Lewin A. Genome Biology, 2011 Feb; 12:R13. doi: 10.1186/gb-2011-12-2-r13.
If you use the mmdiff or mmcollapse programs, please also cite:
- Flexible analysis of RNA-seq data using mixed effects models. Turro E, Astle WJ and Tavaré S. Bioinformatics, 2013. doi: 10.1093/bioinformatics/btt624.
Key features
- Isoform-level expression analysis (works out of the box with Ensembl cDNA and ncRNA files)
- Gene-level expression analysis that is robust to changes in isoform usage, unlike count-based methods
- Haplotype-specific analysis, useful for eQTL analysis and studies of F1 crosses
- Multi-mapping of reads, including mapping to transcripts from different genes, is properly taken into account
- The insert size distribution is taken into account
- Sequence-specific biases can be taken into account
- Flexible differential analysis based on linear mixed models
- Uncertainty in expression parameters is taken into account
- Polytomous model selection (i.e. selecting amongst numerous competing models)
- Modelling of isoform usage proportions
- Collapsing of transcripts with high levels of uncertainty into inferential units which can be estimated with reduced uncertainty
- Multi-threaded C++ implementations
Installation
Download the latest release of MMSEQ, unzip and add the bin directory to your PATH. E.g.:
wget -O mmseq-latest.zip https://github.com/eturro/mmseq/archive/latest.zip
unzip mmseq-latest.zip && cd mmseq-latest
export PATH=`pwd`/bin:$PATHYou might want to strip the suffix from the binaries. E.g., under Linux:
cd bin
for f in `ls *-linux`; do
mv $f `basename $f -linux`
doneThe current release is 1.0.10 (changelog). Visit the release archive to download older releases.
Estimating expression levels
Input files:
- FASTQ files containing reads from the experiment
- A FASTA file containing transcript sequences to align to (find ready-made files)
The example commands below assume that the FASTQ files are asample_1.fq and asample_2.fq (paired-end) and the FASTA file is Homo_sapiens.GRCh37.70.ref_transcripts.fa.
Step 1a: Index the reference transcript sequences with Bowtie 1 (not Bowtie 2); to use kallisto instead, see Step 1b
bowtie-build --offrate 3 Homo_sapiens.GRCh37.70.ref_transcripts.fa Homo_sapiens.GRCh37.70.ref_transcripts (It is advisable to use a lower-than-default value for --offrate (such as 2 or 3) as long as the resulting index fits in memory.)
Step 1b: Index the reference transcript sequences with kallisto (this is an alternative to Step 1a)
kallisto index -i Homo_sapiens.GRCh37.70.ref_transcripts.kind Homo_sapiens.GRCh37.70.ref_transcripts.faStep 1c (optional): Trim out adapter sequences if necessary
If the insert size distribution overlaps the read length, trim back the reads to exclude adapter sequences. Trim Galore! works well. E.g. for libraries prepared using standard Illumina adapters (AGATCGGAAGAGC), run:
trim_galore -q 15 --stringency 3 -e 0.05 --length 36 --trim1 --paired asample_1.fq.gz asample_2.fq.gzStep 2a: Align reads with Bowtie 1 (not Bowtie 2); to use kallisto instead, see Step 2b
bowtie -a --best --strata -S -m 100 -X 500 --chunkmbs 256 -p 8 Homo_sapiens.GRCh37.70.ref_transcripts \
-1 <(gzip -dc asample_1.fq.gz) -2 <(gzip -dc asample_2.fq.gz) | samtools view -F 0xC -bS - | \
samtools sort -n - asample.namesorted- Always specify
-ato ensure you get multi-mapping alignments - Suppress alignments for reads that map to a huge number of transcripts with the
-moption (e.g.-m 100) - Adjust
-Xaccording to the maximum insert size - Specify
--norcif the data were generated following a forward-stranded protocol - If the reference FASTA file doesn't use the Ensembl convention, then also specify
--fullref - The read names must end with /1 or /2, not /3 or /4 (this can be corrected with
awk 'FNR % 4==1 { sub(/\/[34]$/, "/2") } { print }' secondreads.fq > secondreads-new.fq). - If there are multiple FASTQ files from the same library, feed them all together to Bowtie in one go (delimit the FASTQ file names with commas)
- The command above assumes the FASTQ files are gzipped, hence the
gzip -dc - If you are getting many "Exhausted best-first chunk memory" warnings, try increasing
--chunkmbsto 128 or 256. - If the read names contain spaces, make sure the substring up to the first space in each read is unique, as Bowtie strips anything after a space in a read name
- The output BAM file must be sorted by read name.
- With paired-end data, only pairs where both reads have been aligned are used, so might as well use the samtools
0xCfiltering flag as above to reduce the size of the BAM file
Step 2b: Align reads with kallisto (this is an alternative to Step 2a)
kallisto pseudo -i Homo_sapiens.GRCh37.70.ref_transcripts.kind --pseudobam -o kout asample_1.fq.gz asample_2.fq.gz | \
| samtools view -F 0xC -bS - | samtools sort -n - asample.namesortedStep 3: Map reads to transcript sets
bam2hits Homo_sapiens.GRCh37.70.ref_transcripts.fa asample.namesorted.bam > asample.hitsStep 4: Obtain expression estimates
mmseq asample.hits asampleDescription of output:
-
asample.mmseqcontains a table with columns:- feature_id: name of transcript
- log_mu: posterior mean of the log_e expression parameter (use this as your log expression measure)
- sd: posterior standard deviation of the log_e expression parameter
- mcse: Monte Carlo standard error
- iact: integrated autocorrelation time
- effective_length: effective transcript length
- true_length: length of transcript sequence
- unique_hits: number of reads uniquely mapping to the transcript
- mean_proportion: posterior mean isoform/gene proportion
- mean_probit_proportion: posterior mean of the probit-transformed isoform/gene proportion
- sd_probit_proportion: posterior standard deviation of the probit-transformed isoform/gene proportion
- log_mu_em: log-scale transcript-level EM estimate
- observed: whether or not a feature has hits
- ntranscripts: number of isoforms for the gene of that transcript
-
asample.identical.mmseq: as above but aggregated over transcripts sharing the same sequence (these estimates are usually far more precise than the corresponding individual estimates in the transcript-level table); note thatlog_mu_emand proportion summaries are not available for aggregates -
asample.gene.mmseq: as above but aggregated over genes and theeffective_lengthis an average of isoform effective lengths weighted by their expression -
Various other files (
asample.*.trace_gibbs.gz,asample.Mandasample.k) containing more detailed output
These steps operate on a sample-by-sample basis and the expression estimates are roughly proportional to the RNA concentrations in each sample. Some scaling of the estimates may be required to make them comparable across biological replicates and conditions. This can be achieved by reading in the output for multiple samples using the readmmseq() function defined in the mmseq.R R script included in the src/R directory (more information on this function can be found here). The posterior standard deviations capture the uncertainty due to both Poisson counting noise and the additional ambiguity in the mappings between reads and transcripts. The biological variance across samples can only be discerned with the use of biological replicates (see section on differential expression below).
Differential expression analysis
Flexible model comparison using MMDIFF
The mmdiff binary performs model comparison using the posterior summaries (log_mu and sd or mean_probit_proportion and sd_probit_proportion) saved in the MMSEQ tables. The models, indexed by m, are given by
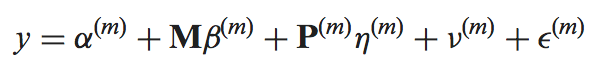
where y is the response (log expression or probit proportion parameter (see below)), M is a model-independent nuisance covariate, P is a model-dependent covariate, "nu" is a random effect capturing the posterior uncertainty in the response and "epsilon" is the residual.
Two models can be specified in a matrices file using the -m option (see below). Alternatively, for standard differential expression analysis (comparison of a model specifying two or more conditions with a model specifying a single condition), the -de convenience option may be used instead of -m. E.g. for a simple 2 vs. 2 gene-level comparison, run:
mmdiff -de 2 2 cond1rep1.gene.mmseq cond1rep2.gene.mmseq cond2rep1.gene.mmseq cond2rep2.gene.mmseq > out.mmdiffThe prior probability that the second model is true may be specified with -p FLOAT (default: 0.1).
In order to use the probit-transformed isoform usage proportions for inference (in order to check for differential isoform usage), set the option -useprops.
The models are regression-based and more complex experimental designs can be specified in a file containing four matrices:
- M is a model-independent covariate matrix
- C maps observations to classes for each model
- P0(collapsed) is a collapsed matrix (i.e. distinct rows) defining statistical model 0
- P1(collapsed) is a collapsed matrix defining statistical model 1
If a model has no classes (i.e. just an intercept term), define the P matrix to be simply 1. E.g. for transcript-level differential expression between two groups of two samples without extraneous variables, the following command and matrices file would be appropriate (note, this is equivalent to specifying -de 2 2):
mmdiff -m matrices_file cond1rep1.mmseq cond1rep2.mmseq cond2rep1.mmseq cond2rep1.mmseq > out.mmdiffwhere the matrices file contains:
# M; no. of rows = no. of observations
0
0
0
0
# C; no. of rows = no. of observations and no. of columns = 2 (one for each model)
0 0
0 0
0 1
0 1
# P0(collapsed); no. of rows = no. of classes for model 0
1
# P1(collapsed); no. of rows = no. of classes for model 1
.5
-.5The resulting design matrices and associated coefficients would be:
# model 0 ([1|P0]):
alpha0
1
1
1
1
# model 1 ([1|P1])
alpha1 eta1_0
1 .5
1 .5
1 -.5
1 -.5Model-independent nuisance covariates can be specified in the M matrix. E.g. in a 3 vs 3 comparison with a single binary nuisance covariate, use:
# M; no. of rows = no. of observations
1
0
1
0
1
0
# C; no. of rows = no. of observations and no. of columns = 2 (one for each model)
0 0
0 0
0 0
0 1
0 1
0 1
# P0(collapsed); no. of rows = no. of classes for model 0
1
# P1(collapsed); no. of rows = no. of classes for model 1
.5
-.5The resulting design matrices and associated coefficients would be:
# model 0 ([1|M|P0]):
alpha0 beta0
1 1
1 0
1 1
1 0
1 1
1 0
# model 1 ([1|M|P1])
alpha1 beta1 eta1_0
1 1 .5
1 0 .5
1 1 .5
1 0 -.5
1 1 -.5
1 0 -.5For a three-way differential expression analysis with three, three and two observations per group respectively, this matrices file would be appropriate (equivalent to using -de 3 3 2).
In order to assess whether the log fold change between group A and group B is different (as opposed to equal) to the log fold change between group C and group D, assuming there are two observations per group, this matrices file would be appropriate.
By default, mmdiff includes a global intercept alpha. If you prefer to fix alpha=0 and instead use the M covariate matrix to define multiple independent intercepts (beta), then set the option -fixalpha. This can be used to obtain class-specific expression summaries without performing model comparison: specify a prior probability of either model to zero (use -p 0), remove the intercept term using -fixalpha and use the M matrix to group samples into groups using a matrices file such as the following, which groups samples into sets of two:
# M; no. of rows = no. of observations
1 0 0
1 0 0
0 1 0
0 1 0
0 0 1
0 0 1
# C; no. of rows = no. of observations and no. of columns = 2 (one for each model)
0 0
0 0
0 0
0 0
0 0
0 0
# P0(collapsed); no. of rows = no. of classes for model 0
1
# P1(collapsed); no. of rows = no. of classes for model 1
1The resulting design matrix and associated coefficients would then be:
# model 0 ([M]):
beta0_0 beta0_1 beta0_2
1 0 0
1 0 0
0 1 0
0 1 0
0 0 1
0 0 1Thus, the posterior summaries encoded in beta0_0, beta0_1, beta0_2 would represent the class-specific expression estimates.
For further advice on setting up the matrices file for a particular study design, do not hesitate to contact the corresponding author.
Description of the output:
- feature_id: the name of the feature (e.g. Ensembl transcript ID)
- bayes_factor: the Bayes factor in favour of the second model
- posterior_probability: the posterior probability in favour of the second model (the prior probability is recorded in a # comment at the top of the file)
- alpha0 and alpha1: estimated posterior mean of the global intercept for each model
- beta0_0, beta0_1..., beta1_0, beta1_1...: estimated posterior means of the regression coefficients of the model-independent covariate matrix M under each model
- eta0_0, eta0_1..., eta1_0, eta1_1...: estimated posterior means of the regression coefficients of the model-dependent matrix P under each model
- mu_sample1, mu_sample2,... sd_sample1, sd_sample2,...: the data, i.e. the posterior means and standard deviations used as the outcomes
To recompute the posterior probability based on a different prior probability without having to re-run mmdiff, you can use the following function in R, where bf is the Bayes factor and prior is the new desired prior probability:
recompute_pp <- function(bf, prior) { 1/(1 + exp( -( log(bf) + log(prior) - log(1-prior) ))) }To perform polytomous model selection (i.e., compare multiple models), run mmdiff multiple times, comparing each alternative model to some baseline model. Then, in R, source the mmseq.R file in the src/R directory and feed the mmdiff output files to the polyclass() function. A vector of prior probabilities for each model can be specified in the prior argument (assumed flat by default). The function returns a data frame containing the posterior probability of each model for each feature (postprob_model0, postprob_model1, etc.).
For an alternative approach to differential expression analysis using edgeR or DESeq, view these instructions.
Collapsing transcripts
It is possible to collapse sets of transcripts based on posterior correlation estimates with the mmcollapse binary. The overall precision of the expression estimates for a collapsed set is usually much greater than for the individual component transcripts, which typically share important structural features (many shared exons, etc). The mmcollapse binary uses the output of mmseq to do the collapsing:
mmcollapse basename1 basename2...Here, basename1 basename2... corresponds to the name of the mmseq output files without the suffixes (such as .mmseq and .gene.mmseq). Pass all the basenames from the same experiment to mmcollapse in one go to ensure the collapsings are consistent across samples. The command creates a set of new transcript-level MMSEQ files with the suffix .collapsed.mmseq, which can be used to perform more powerful transcript-level differential analysis.
Reference files
Ready to download:
- Homo sapiens: download transcriptome FASTA files containing cDNA and ncRNA transcript sequences (but excluding alternative haplotype/supercontig entries) for the following versions of Ensembl: 64, 68, 70.
- Mus musculus: genome and transcriptome FASTA files based on the GRCm38 build and the March 2013 SNP and indel calls from the Wellcome Trust Mouse Genomes Project are available here for the following strains: C57BL6, 129P2, 129S1, 129S5, AJ, AKRJ, BALBcJ, C3HHeJ, C57BL6NJ, CASTEiJ, CBAJ, DBA2J, FVBNJ, LPJ, NODShiLtJ, NZOHlLtJ, PWKPhJ, SPRETEiJ and WSBEiJ. These files were generated using the mouse_strain_transcriptomes.sh script. Please cite the papers if you publish work using these files. For F1 data, append "_STRAIN" to each transcript and gene ID in the transcriptome FASTA headers (where "STRAIN" is the name of the strain) and concatenate the two relevant files into one hybrid FASTA. Then align the F1 reads to the hybrid reference as per the documentation above. For analysis with
mmdiff, the*.mmseqfiles should be split into two, one file for each strain (useheadto extract the headers andgrep STRAINto extract the rows for a particular strain).
Making your own Ensembl vertebrate reference FASTAs:
Download the cDNA and ncRNA FASTA files for the Ensembl version and species of interest from the Ensembl FTP server and combine them into a single file. For humans, remove alternative haplotype/supercontig entries using the fastagrep.sh script in the bin directory, which is an adapted version of unix grep that greps multi-line FASTA entries. E.g.:
wget ftp://ftp.ensembl.org/pub/release-72/fasta/homo_sapiens/cdna/Homo_sapiens.GRCh37.72.cdna.all.fa.gz
wget ftp://ftp.ensembl.org/pub/release-72/fasta/homo_sapiens/ncrna/Homo_sapiens.GRCh37.72.ncrna.fa.gz
gunzip Homo_sapiens.GRCh37.72.*.fa.gz
cat Homo_sapiens.GRCh37.72.ncrna.fa >> Homo_sapiens.GRCh37.72.cdna.all.fa
fastagrep.sh -v 'supercontig|GRCh37:[^1-9XMY]' Homo_sapiens.GRCh37.72.cdna.all.fa > Homo_sapiens.GRCh37.72.ref_transcripts.fa # for humans only
rm Homo_sapiens.GRCh37.72.cdna.all.fa Homo_sapiens.GRCh37.72.ncrna.fa # clean upFor mice, you may need to remove patches and unscaffolded contigs. E.g.:
wget ftp://ftp.ensembl.org/pub/release-75/fasta/mus_musculus/cdna/Mus_musculus.GRCm38.75.cdna.all.fa.gz
wget ftp://ftp.ensembl.org/pub/release-75/fasta/mus_musculus/ncrna/Mus_musculus.GRCm38.75.ncrna.fa.gz
gunzip Mus_musculus.GRCm38.75.*.fa.gz
cat Mus_musculus.GRCm38.75.ncrna.fa >> Mus_musculus.GRCm38.75.cdna.all.fa
fastagrep.sh -v 'scaffold|PATCH' Mus_musculus.GRCm38.75.cdna.all.fa > Mus_musculus.GRCm38.75.ref_transcripts.fa # for mice only
rm Mus_musculus.GRCm38.75.cdna.all.fa Mus_musculus.GRCm38.75.ncrna.fa # clean upFASTAs with other header conventions:
MMSEQ works out of the box with Ensembl FASTA files, which follow the convention >TID ... gene:GID ..., where TID is the Ensembl transcript ID, GID is the Ensembl gene ID, and the ... may contain other information, such as chromosomal location. In order to use FASTA files that follow a different convention, tell bam2hits how to extract the transcript and gene IDs from the FASTA header strings using the -m tg_regexp t_ind g_ind option. tg_regexp specifies a regular expression that matches FASTA entries (excluding the leading >), where pairs of brackets are used to identify the location of the transcript and gene ID strings. t_ind and g_ind are 1-based indexes to indicate which pair of brackets capture the transcript and gene IDs respectively. For example, if your entries looked like this:
>transcript1234 gene567|chr1|1234then run bowtie with --fullref and run bam2hits with -m "(\S+) ([^\|]+)\|.*" 1 2.
If you are unsure what regular expression to use, try out by trial and error using the testregexp.rb script in the bin directory. After running bam2hits with the -m option, run hitstools inspect hits_file and compare the header and the main lines to make sure the extraction of IDs worked as expected.
Making sample-specific transcript FASTAs through genotyping and phasing:
If you have phased genotypes and wish to create a sample-specific transcript FASTA file, you can use the haploref.rb script. Suppose there was a two-exon transcript called ENST123 on chromosome 1 with reference sequence ATCGAATGA spread across two exons at positions 1000-1003 and 1010-1014. Suppose a particular individual carried G and T on one haplotype and C and G on the other haplotype, at positions 1003 and 1011 of chromosome 1, respectively. This information should be encoded in four files, as follows:
- A reference transcript FASTA (
transcriptome.fa)>ENST123 gene:ENSG123 ATCGAATGA - An Ensembl-style GFF file (
annotations.gff)1 protein_coding mRNA 1000 1014 . + . ID=ENST123;Name=ABCD 1 protein_coding exon 1000 1003 . + . ID=ENST123.1;Name=ABCD;Parent=ENST123 1 protein_coding exon 1010 1014 . + . ID=ENST123.2;Name=ABCD;Parent=ENST123 - A position file (
pos_file)ENST123 1 1003 1011 - A haplotype file (
hap_file)ENST123_A GT ENST123_B CGThe
haploref.rbcommand can then be run to generate a sample-specific FASTA file as follows (redirect stdout to a file):haploref.rb transcriptome.fa annotations.gff pos_file hap_file 2> /dev/null >ENST123_A gene:ENSG123_A ATCGATTGA >ENST123_B gene:ENSG123_B ATCCAGTGABuilding from source
The MMSEQ package comes with statically-linked binaries for 64-bit Mac OS X and GNU/Linux, which should work out of the box on most systems. However, due to a lack of OpenMP support on Apple's clang compiler (as of El Capitan), the Mac binaries are single-threaded. In order to build from source, install the following dependencies (more info):
- Boost C++ libraries
- GNU Scientific Library
- Armadillo C++ linear algebra library (with lapack and blas)
- HTSlib library
Then clone the GitHub repository and run make from the src directory, which will place the binaries in the bin directory:
git clone https://github.com/eturro/mmseq.git
cd mmseq/src
makeMiscellany
-
Parallelism: the number of threads spawned by
bam2hits,mmseqandmmdiffcan be controlled by setting theOMP_NUM_THREADSenvironment variable. The value can be set immediately preceeding the command, e.g.OMP_NUM_THREADS=8 mmseq sample.mmseq sample. -
The
.hitsfiles contain a header section and a main section. The header contains three metadata sections with lines beginning with@:@TranscriptMetaData: information about each of the transcripts. Each line contains transcript ID, effective transcript length, true transcript length@GeneIsoforms: grouping of transcripts into genes. Each line contains gene ID, transcript ID 1, transcript ID 2, ...@IdenticalTranscripts: grouping of transcripts into identical sets. Each line contains transcript ID 1, transcript ID 2, ...
The remainder of the .hits file links each read with a set of transcripts. E.g.:
>read1
transcriptA
transcriptB
>read2
transcript C
...The full file can be printed out with the command hitstools t hits_file (see below).
Software usage
See authors.
bam2hits
Usage: bam2hits [-v] [-t] [-m tg_regexp t_ind g_ind] [-i expected_isize sd_of_isizes] [-k] [-b] [-c alt_lengths] [-u alt_lengths] transcript_fasta sorted_bam > hits_file
Generate hits file for use with `mmseq`.
Mandatory arguments:
transcript_fasta: FASTA file used as reference to produce SAM file.
sorted_bam: BAM file sorted by read name containing alignments to reference transcripts.
Option -v: print version and quit.
Option -t: output hits file in plain text instead of compressed binary format.
Arguments to option -m for use with non-Ensembl transcript FASTA files:
tg_regexp: regular expression matching FASTA entry names, where pairs of brackets
are used to capture transcript and gene IDs. E.g.: "(\S+).*gene:(\S+).*"
t_ind: index of bracket pair that captures the transcript ID. E.g.: 1.
g_ind: index of bracket pair that captures the gene ID. E.g.: 2.
Arguments to option -i for calculating effective transcript lengths and filtering very unlikely alignments:
expected_isize: expected insert size (e.g. 180). Default: for paired-end reads, the mode obtained using the
first 2M single-mapping proper pairs, smoothing the histogram in windows of 3;
for single-end reads, 180.
sd_of_isizes: standard deviation of insert sizes (e.g. 25). Default: for paired-end reads, 1/1.96 times
the distance between the mode and the top 2.50% insert sizes in the first 2M single-mapping proper pairs;
for single-end reads, 25.
Option -k: for paired-end reads, disable insert size deviation filter, which removes alignments with
insert sizes beyond 1.96 sd of the mean if alternative alignments exist
with insert sizes within 1.96 sd of the mean.
Option -b: merge hits to _A and _B arbitrary-phase haplo-isoforms. This is usually done if the only
reason for creating haplo-isoform sequences is to reduce allelic mapping biases.
Argument to option -c for adjusting transcript lengths for non-uniformity with 'mseq' R package (required):
alt_lengths: name of file in which to save the adjusted lengths (can be reused in future with -u)
Argument to option -u for correcting for non-uniformity:
alt_lengths: name of file containing adjusted transcript lengthshitstools
Utilities for handling hitsfile (.hits) and bitsfile (.bits, `binary hitsfile`) formats.
View a summary of a hits file:
hitstools inspect in.hits
Output header:
hitstools header in.hits
Convert from text format to binary format:
hitstools b in_text.hits > out_binary.hits
Convert from binary format to text format:
hitstools t in_binary.hits > out_text.hitsmmseq
Usage: mmseq [OPTIONS...] hits_file output_base
Mandatory arguments:
hits_file hits file generated with `bam2hits`
output_base base name for output files
Optional arguments:
-alpha FLOAT value of alpha in Gamma prior for mu (default: 0.1)
-beta FLOAT value of beta in Gamma prior for mu (default: 0.1)
-max_em_iter INT maximum number of EM iterations (default: 1000)
-epsilon FLOAT minimum loglik ratio between successive EM iterations (default: 0.1)
-gibbs_iter INT number of Gibbs iterations (default: 16384)
-gibbs_ss INT subsampling interval for Gibbs output (default: gibbs_iter/1024)
-seed INT seed for the PRNG in thread 0 (default: 1234)
-debug output additional diagnostic files
-help print this help message
-version print the versionmmdiff
Usage: mmdiff [OPTIONS...] [-de n1 n2 ... nC | -m matrices_file] mmseq_file1 mmseq_file2... > out.mmdiff
matrices_file contains M P0 P1 each separated by an empty line
Mandatory arguments:
ONE OF:
-de INT INT... simple differential expression between several groups of samples, where
each INT corresponds to a grouping of MMSEQ files into one condition
-m STRING path to matrices file specifying the two models to compare
Optional arguments:
-tracedir STRING directory in which to save MCMC traces (default: (do not write traces))
-useprops run on isoform/gene proportions instead of expression
-permute run on permuted dataset; combine with non-permuted results to obtain q-values
-p FLOAT prior probability of the second model (default: 0.1)
-d FLOAT d hyperparameter (default: 1.4)
-s FLOAT s hyperparameter (default: 2.0)
-l INT length of MCMC trace used to produce the input estimates (default: 1024)
-fixalpha fix alpha=0 and do not update (default: estimate alpha)
-nonorm do not normalise the input data
-pdash FLOAT initial value of pdash for improved mixing of gamma (stays fixed if -notune is set) (default: 0.5)
-notune do not tune pdash to improve mixing for gamma
-uhfrac FLOAT if normalising, use features which have at least one unique hit
in at least uhfrac of the samples (default: max(0.2, (N - floor(N^2/160))/N))
-burnin INT burnin iterations (default: 8192)
-iter INT MCMC iterations (default: 16384)
-seed INT seed for PRNG (default: 1234)
-range INT INT select features indexed within range (default: all)mmcollapse
Usage: mmcollapse basename1 basename2...extract_transcripts
Usage: extract_transcripts fasta_file ensembl_gtf_fileoffsetGTF
Usage: offsetGTF ensembl_gtf_file indel_vcf_filet2g_hits
Usage: t2g_hits <(hitstools t hits_file) > gene_hits_file
Convert a transcript-level hits file in text format into a gene-level hits file in text format
Mandatory arguments:
hits_file hits file generated with `bam2hits`fastagrep.sh
Usage: fastagrep.sh: [-v] pattern fasta_filetestregexp.rb
Usage: testregexp.rb -m tg_regexp t_ind g_ind cdna_file
Test regular expression patterns for use with `bam2hits` and `haploref.rb`.
Arguments to flag -m:
tg_regexp: regular expression matching FASTA entry names, where pairs of brackets
are used to capture transcript and gene IDs. Default: "(\S+).*gene:(\S+).*"
t_ind: index of bracket pair that captures the transcript ID. Default: 1.
g_ind: index of bracket pair that captures the gene ID. Default: 2.
cdna_file: reference FASTA file.filterGTF.rb
Usage: filterGTF.rb cdna_file gtf_file > new_gtf_file
Filter GTF file according to transcripts in cDNA file.
Mandatory arguments:
cdna_file: Ensembl transcript FASTA file (e.g. Homo_sapiens.GRCh37.56.cdna.ref.fa).
gtf_file: Ensembl GTF file (e.g. Homo_sapiens.GRCh37.56.gtf).haploref.rb
Usage: haploref.rb [-m tg_regexp t_ind g_ind] cdna_file gff_file pos_file hap_file > hapiso_fasta
Generate haplotype and isoform specific FASTA reference.
Mandatory arguments:
cdna_file: default transcript FASTA file.
gff_file: GFF file containing structure annotation for transcripts in cdna_file.
pos_file: file containing, for each transcript, chromosome and positions of SNPs.
hap_file: file containing, for each transcript, two versions, e.g. suffixed _A and _B, on separate lines with the alleles for the two haplotypes at each position listed in the pos_file (A and B respectively).
Arguments to option -m for use with non-Ensembl cDNA FASTA files:
tg_regexp: regular expression matching FASTA entry names, where pairs of brackets
are used to capture transcript and gene IDs. Default: "(\S+).*gene:(\S+).*"
t_ind: index of bracket pair that captures the transcript ID. Default: 1.
g_ind: index of bracket pair that captures the gene ID. Default: 2.ensembl_gtf_to_gff.pl
convert gtf file from Ensembl to gff3 file
Usage: ensembl_gtf_to_gff.pl gtf_file > gff_file