Behavioral Guided Transcriptomics (BGT) Pipeline
BGT is an imaging-guided transcriptomic experimental and analytical pipeline for co-cultures of Patient Derived Organoides with (engeneered) T cells described in Dekkers&Alieva et al Nat. Biotech (2022). This repository compiles the analytical procedures required to integrate data from single-cell RNA sequencing of T cells with single-cell imaging data, providing for each sequenced cell probabilities of exhibiting different functional behaviors.
Cloning Instructions
To clone this repository to your local machine, you will need Git installed. Follow these instructions to install Git if you haven't already.
Once Git is installed, open your terminal (Command Prompt or PowerShell on Windows, Terminal on macOS and Linux) and run the following command:
git clone https://github.com/AlievaRios/BGT.gitInput data
Imaging-derived dataset of behavior-classified T cells.
This data is obtained by performing multispectral time-lapse imaging of T cells incubated with tumor organoids and processed with BEHAV3D. Refer to BEHAV3D repository and our detailed Protocol to obtain this data Alieva et al, Nat Protoc, 2024. User needs to run Module 2. T cell Behavior Classification of BEHAV3D to generate "classified_tcell_track_data.rds" file which is directly utilized as an input for Modules 1. Evaluation of super-engager population dynamics in co-culture and Module 3. in silico population separation simulation of the BGT pipeline.
Single-cell sequencing data from T cells incubated with patient-derived organoids.
This data is obtained from running SORT seq on T cells incubated with tumor organoids. The scRNA seq file we provide for the demo is 10T_master.rds which you can find under the data folder
Extended Software and Hardware Requirements
As the BGT pipeline is an extended implementation to the BEHAV3D protocol, the input data required to apply it is generated from BEHAV3D pipeline, which has been extensively described Alieva et al, Nat Protoc, 2024 BEHAV3D demands high-end workstations (RAM 64GB, CPU 3.7 HZ or more with at least 8 cores, GPU with 16GB VRAM or more) for data processing with Imaris ensuring analysis efficiency. For applying the BGT data analysis pipeline, a general workstation will be sufficient. In terms of user expertise, a basic understanding of R programming is required to run the data analysis pipeline.
Installations
You have two options to execute the pipeline:
- Option one (Recommended): using the provided Singularity image. This pre-configured image includes all necessary dependencies, reducing manual installation steps and potential errors. This installation was tested in Windows and Linux.
- Option two: Installing all the libraries and packages yourself (Make sure to use the specific versions of the packages listed)
OPTION 1
Install WSL: a Linux Distribution (For Windows Users)
Open a PowerShell command line (search in the Windows bar for PowerShell) and execute this command Note: Skip to step 1 if you already have Wsl or linux as the operating system in your analysis PC.
wsl --install -d Ubuntu-22.04Note: After this step, it is recommended to reboot your system.
Follow the Guide from Singularity Tutorial OR the following steps
If you are not using Ubuntu or something in the tutorial below doesn't work, refer to the official guide at Singularity Tutorial.
1. Install Dependencies
Open a terminal in Linux (in Windows, go to the search bar and type Ubuntu) and paste the commands one by one:
sudo apt-get updatesudo apt-get install -y build-essential libssl-dev uuid-dev libgpgme11-dev \
squashfs-tools libseccomp-dev wget pkg-config git cryptsetup debootstrap2. Install Go
Paste the commands one by one:
wget https://dl.google.com/go/go1.13.linux-amd64.tar.gzsudo tar --directory=/usr/local -xzvf go1.13.linux-amd64.tar.gzexport PATH=/usr/local/go/bin:$PATH3. Install Singularity
Paste the commands one by one:
wget https://github.com/singularityware/singularity/releases/download/v3.5.3/singularity-3.5.3.tar.gztar -xzvf singularity-3.5.3.tar.gzcd singularity./mconfigcd builddirmakesudo make install4. Configure Bash Completion for Singularity
Paste the commands one by one:
source etc/bash_completion.d/singularitysudo cp etc/bash_completion.d/singularity /etc/bash_completion.d/5. Clone your BGT Repository
git clone https://github.com/AlievaRios/BGT.git6. Download your Image
Download the Singularity image Link .
7. Execute Your Image
Once you have everything installed, proceed to execute the image you downloaded.
singularity shell --pid bgt_image.sif8. Start RStudio Server
When you are inside the Singularity image, execute this command:
rstudio-server start9. Enter RStudio
Copy and paste http://localhost:8787 this webpage URL. The webpage will be accessible through any web browser of your choice. You can now run BGT Demo from the Rstudio, instructions to which are provided here.
NOTE: Rstudio will be closed if you kill the terminal (the data will be preserved)
OPTION 2
Install all the following libraries (R version 4.3.2 (2023) and 4.0.5 (2021) :
| Package | Version |
|---|---|
| abind | 1.4-5 |
| dplyr | 1.1.4 |
| dtwclust | 5.5.12 |
| fs | 1.6.3 |
| future | 1.33.1 |
| furrr | 0.3.1 |
| ggplot2 | 3.4.4 |
| gplots | 3.1.3 |
| MESS | 0.5.9 |
| optparse | 1.7.3 |
| parallel | 4.3.0 |
| patchwork | 1.2.0 |
| pheatmap | 1.0.12 |
| plyr | 1.8.8 |
| randomForest | 4.7-1.1 |
| readr | 2.1.4 |
| reshape2 | 1.4.4 |
| scales | 1.3.0 |
| Seurat | 5.0.1 |
| SeuratObject | 5.0.1 |
| spatstat | 3.0-6 |
| sp | 1.6-1 |
| stats | 4.3.0 |
| tibble | 3.2.1 |
| tidyr | 1.3.0 |
| tidyverse | 2.0.0 |
| umap | 0.2.10.0 |
| viridis | 0.6.4 |
| viridisLite | 0.4.2 |
| xlsx | 0.6.5 |
| yaml | 2.3.8 |
| zoo | 1.8-12 |
| BiocManager | 3.18 |
| ggthemes | (latest) |
| gridExtra | (latest) |
| openxlsx | (latest) |
| devtools | (latest) |
| monocle3 | (latest) |
Note: For packages listed with '(latest)' as their version, the user should install the most recent version available at the time of installation. This can be done using the install.packages() function for CRAN packages and the BiocManager::install() function for Bioconductor packages, without specifying a version number.
Repository Structure
This repository contains a collection of scripts and example datasets enabling the following dowstream analysis. Follow the structure in the script folder for each module and each analysis type. Introduce the corresponding folder/ file direction on your own computer where required (note that to specify directory paths in R (/) forward slash is recommended):
Demos
You can run BGT on demo data to see examples of the results. This will take <20 minutes
Modules

(1) Evaluation of optimal timing for ‘Super engaged’ enrichment
This module analyzes T-cell engagement dynamics, particularly highlighting the activity within cluster 9—representative of super-engagers. It enables a detailed comparison between CD4 and CD8 T-cells' engagement over time in co-culture experiments, visualizing the engagement percentage of T-cells in the super-engager state across various time points. This analysis marks the time window during which the super-engager population is most discernible, aiding in the accurate prediction of the timepoint for T cell separation. The classified_tcell_track_data.rds file, encompassing the classified behavioral data for T cells, and the determined starting point of imaging (imaging time), is critical for this analysis. These parameters should be incorporated into the provided BGT_config template within the simulation settings section. These must be accurate and representative of your specific experimental setup.
Configuration
Before running the module, ensure the config file (config_template.yml) is correctly set up with your specific parameters, including:
classified_tcell_track_data_filepath_rds: Path to the "classified_tcell_track_data.rds" file.imaging_time: Time (in hours) when imaging starts in the experiment; this module will focus on time points after this.
To run from the command line:
Rscript /path/to/TimepointGraph/Timepoint_graph.R -c </Path/to/BGT/config_template.yml> -fThe -f flag forces the re-import and processing of data even if the output files already exist. Optionally, use -t to specify an RDS file containing the processed "classified_tcell_track_data.rds" file, if not already included in the BGT config.
To run from RStudio:
Step 1: use the T cell behavioral dynamics for a demo run. Adjust the path to your BGT config on line 18 if you're using a different data folder or config file.
Output Files
Outputs are saved in the specified output_dir within the Results/Seperation_time_graph/ directory:
- T-cell_engagementVsTime.png: Visualization of the percentage of T cells in cluster 9 (super-engagers) over time.
- T-cell_engagementVsTime.csv: Raw data file containing the time points and percentages of T cells in cluster 9, ready for further analysis.
Key Features
- Temporal Dynamics Insight: The module reveals critical engagement dynamics, identifying when super-engager T cells are most prevalent.
- Cluster 9 Focus: By concentrating on cluster 9, it provides an in-depth look at the behavior of T cells demonstrating high engagement levels.
- CD4 vs. CD8 Comparison: Offers valuable insights into distinct engagement patterns between CD4 and CD8 T cells, enhancing the understanding of immune mechanisms.
For additional analysis or adjustments to visualization parameters, consult the script. This might include altering smoothing functions or the analysis time window, depending on the specifics of your experimental setup.
(2) scRNAseq data preprocessing
This module contains scripts to process and analyze single-cell sequencing data of T cells. To test this pipeline you can either use the provided demos datasets or download an example sequencing data deposited in the GEO depository with the accession number GSE172325. You have two ways of running these scripts: through Jupyter Notebooks or Markdown R scripts.
To run from RStudio:
| Step 1: For a demo run, use these scripts. Adjust the path to your data folder inside each script (at the begining of each file). This module is structured in the following way: | Script | Jupyter Notebook | R Markdown | Key Features |
|---|---|---|---|---|
| 1 - Pre-processing of raw SORTseq data | 1 Script | 1 Script | This script is for an initial, per-plate (visual) inspection and to prepare expression matrices for the downstream steps. | |
| 2 - Subset Identification | 2 Script | 2 Script | This script aims to identify different T cell subsets in the dataset and annotate them. | |
| 3 - Pseudotime Analysis | 3 Script | 3 Script | This script aims to infer pseudotime trajectory for TEG subsets we identified during the previous analysis pipeline. | |
| 4 - Dynamic Gene Clustering | 4 Script | 4 Script | This script aims to cluster dynamic genes along the trajectory (identified as the result of running the previous part of the whole analysis pipeline) into categories of genes with highly similar behavior. | |
| 5 - Comparison to In vivo Data | 5 Script | 5 Script | This script aims to identify marker genes related to highly tumor-reactive T cells in cancer tumor microenvironment datasets and check whether those genes are also enriched in our TEG dataset's clusters. |
(3) In silico population separation simulation
This module simulates T-cell population separation into engagers and non-engagers over multiple time points, providing insights into T-cell dynamics in a co-culture environment. It utilizes user-defined parameters from the configuration file to analyze and visualize the engagement behavior of CD4 and CD8 T-cells.
Configuration
Set up the config_template.yml with the necessary parameters for this module, including:
classified_tcell_track_data_filepath_rds: Path to the "classified_tcell_track_data.rds" file.n_timepoints: The number of separation time points to include in the simulation.first_timepoint_interval_startsandfirst_timepoint_interval_ends: The starting and ending minutes of the first time interval for separation.reoccuring_time: Time difference (in minutes) for subsequent time intervals.
To run from the command line:
Rscript /path/to/PopulationSeparationSimulation/Population_separation_simulation.R -c </Path/to/BGT/config_template.yml> -fUse the -f flag to force re-import and processing of data if output files already exist. The -t option allows specifying an RDS file with processed T-cell track data, if not included in the BGT config.
To run from RStudio:
Step 2: For a demo, navigate to the population_separation_simulation script. Adjust the BGT config file path on line 18) if working with new data in a different folder.
Output Files
Generated outputs in the Results/Population_seperation_simulation/ directory include:
- Plots for CD8 and CD4 T-cell engagement at specified intervals, indicating the percentage of cells in different clusters.
plot_cd8_timepoint_X.pngandplot_cd4_timepoint_X.pngwhere X denotes the specific time interval.
- CSV files detailing the engagement frequency analysis for CD8 and CD4 T-cells:
CD8_engagement_behavior_freq.csvandCD4_engagement_behavior_freq.csv, providing a quantitative measure of T-cell engagement over time.
Key Features
- Dynamic Analysis: Enables a comprehensive view of T-cell behavior across several time points, facilitating the understanding of engagement dynamics.
- Flexible Time Points: Leverages user-defined intervals to tailor the analysis to specific experimental setups, enhancing relevance to your research.
- Detailed Visualization: Offers clear, visual representations of T-cell engagement, aiding in the identification of patterns and trends in cellular behavior.
Adjustments to the analysis parameters or visualization aspects can be made by modifying the script or the config file, depending on the specifics of the experimental design or analysis goals.
(4) Behavioral probability mapping module
This module uses the principle of probability transitivity to infer for each sequenced T cell a probability to belonging to a particular T cell beahvioral class.
Behavioral probability mapping module uses as input processes Seurat object from scRNA analysis module along with Tcell_engagement_behavior_freq.csv from the in silico population separation simulation module.
Configuration
Ensure the config_template.yml is correctly configured for this module:
scRNA_seq_dataset: Path to the single-cell RNA sequencing dataset file derived as output of Module 2.CD8_engagement_behavior_freq.csvandCD4_engagement_behavior_freq.csvfiles detailing the engagement frequency analysis for CD8 and CD4 T-cells derived as output of Module 3- Other parameters relevant to this module are set directly within the script.
To run from the command line:
Rscript /path/to/BehavioralGuidedTranscriptomics/Behavioral-guided_transcriptomics.R -c </Path/to/BGT/config_template.yml> -fUse -f to force re-import and processing of data if output files already exist. Optionally, -t can specify an RDS file with processed T-cell track data not included in the BGT config.
To run from RStudio:
Step 3: For demonstration purposes, utilize the Behavioral probability mapping script. If using new data or a different configuration, update the BGT config file path on line 21.
Output Files
The module outputs include:
- UMAP plots illustrating the distribution of behavioral signatures across the scRNA-seq dataset.
- Probability maps and CSV files detailing the behavioral signature composition for each analyzed cluster.
- Heatmaps and PDFs visualizing the transition of T-cell behaviors along the pseudotime trajectory.
Outputs are stored in Results/Behavioral_guided_transcriptomics/ and its subdirectories.
Key Features
- Behavioral Signature Mapping: Elucidates the transcriptomic underpinnings of T-cell behaviors, offering a nuanced view of immune cell dynamics.
- Probability Maps: Constructs detailed maps showing the likelihood of each cell belonging to different behavioral categories, enabling a deep dive into cellular function and heterogeneity.
- Pseudotime Analysis: Integrates pseudotime analysis to explore the evolution of T-cell states and behaviors over time, revealing insights into cellular progression and differentiation in response to tumor exposure.
For modifications or further analysis, refer to the script and adjust parameters or visualization settings as needed based on your specific research questions or experimental design.
Example output dataset
To enable users to delve into an illustrative dataset generated with BGT, we offer an example showcasing CD8+ TEGs co-cultured with Breast Cancer Organoids, accessible via an online UCSC browser. For information on how to use it go to this page
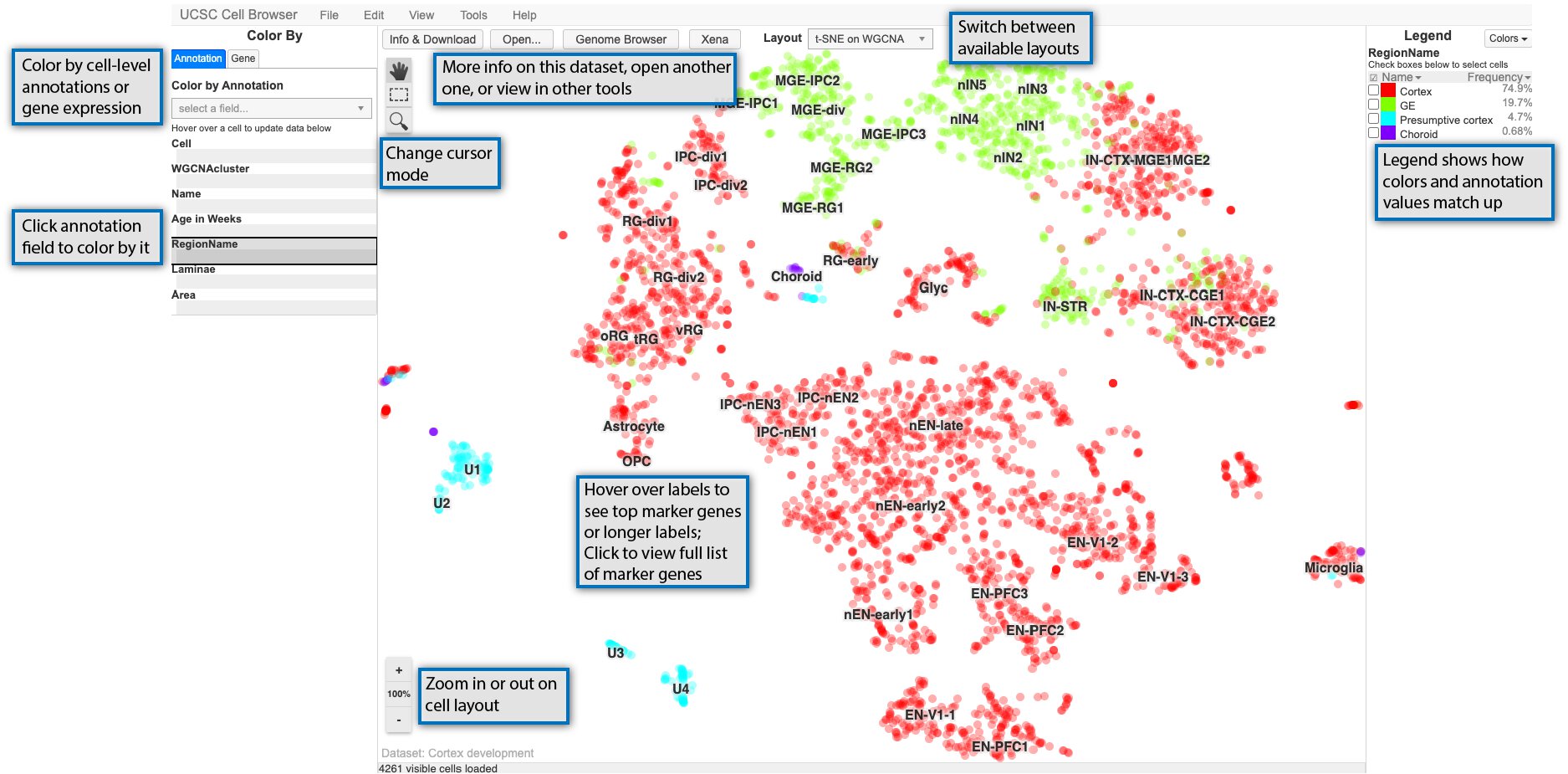
Currently you can view it by :
- Cell
- Oganoid
- Experiment
- Plate
- Pseudotime
- Experimental Condition
- PT Cluster