De novo peptide sequencing with InstaNovo
![]()
![]()

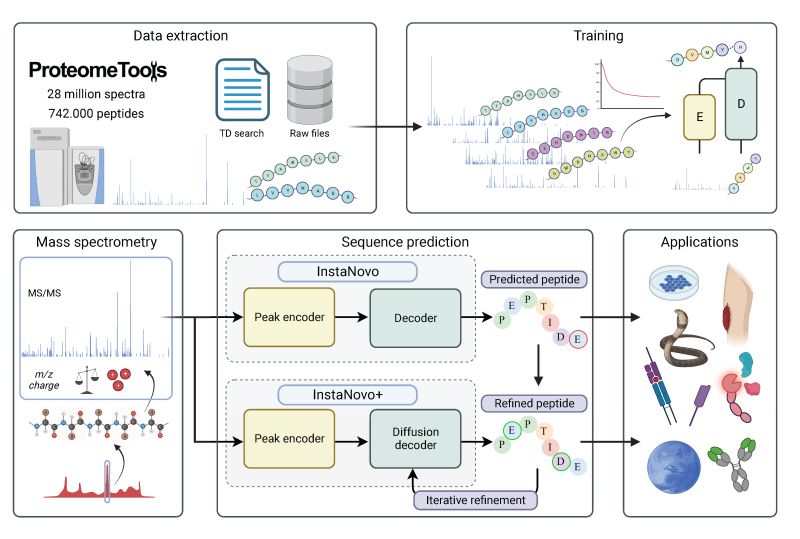
The official code repository for InstaNovo. This repo contains the code for training and inference of InstaNovo and InstaNovo+. InstaNovo is a transformer neural network with the ability to translate fragment ion peaks into the sequence of amino acids that make up the studied peptide(s). InstaNovo+, inspired by human intuition, is a multinomial diffusion model that further improves performance by iterative refinement of predicted sequences.
Links:
- bioRxiv: https://www.biorxiv.org/content/10.1101/2023.08.30.555055v3
- documentation: https://instadeepai.github.io/InstaNovo/
Developed by:
Usage
Installation
To use InstaNovo, we need to install the module via pip:
pip install instanovoIt is recommended to install InstaNovo in a fresh environment, such as Conda or PyEnv. For example, if you have conda/miniconda installed:
conda env create -f environment.yml
conda activate instanovoNote: InstaNovo is built for Python >= 3.10, <3.12 and tested on Linux.
Training
To train auto-regressive InstaNovo using Hydra configs (see --hydra-help for more information):
usage: python -m instanovo.transformer.train [--config-name CONFIG_NAME]
Config options:
config-name Name of Hydra config in `/configs/`
Defaults to `instanovo_acpt`Note: data is expected to be saved as Polars .ipc format. See section on data conversion.
To update the InstaNovo model config, modify the config file under configs/instanovo/base.yaml
Prediction
To get de novo predictions from InstaNovo:
Usage: python -m instanovo.transformer.predict [--config-name CONFIG_NAME] data_path=path/to/data.mgf model_path=path/to/model.ckpt output_path=path/to/output.csv denovo=True
Predict with the model.
Options:
data_path Path to dataset to be evaluated. Must be specified
in config or cli. Allows `.mgf`, `.mzxml`, a directory,
or an `.ipc` file. Glob notation is supported: eg.:
`./experiment/*.mgf`
model_path Path to model to be used. Must be specified
in config or cli. Model must be a `.ckpt` output by the
training script.
output_path Path to output csv file.
config-name Name of Hydra config in `/configs/inference/`
Defaults to `default`To evaluate InstaNovo performance on an annotated dataset:
Usage: python -m instanovo.transformer.predict [--config-name CONFIG_NAME] data_path=path/to/data.mgf model_path=path/to/model.ckpt denovo=False
Predict with the model.
Options:
data_path Path to dataset to be evaluated. Must be specified
in config or cli. Allows `.mgf`, `.mzxml`, a directory,
or an `.ipc` file. Glob notation is supported: eg.:
`./experiment/*.mgf`
model_path Path to model to be used. Must be specified
in config or cli. Model must be a `.ckpt` output by the
training script.
config-name Name of Hydra config in `/configs/inference/`
Defaults to `default`The configuration file for inference may be found under /configs/inference/default.yaml
Note: the denovo=True/False flag controls whether metrics will be calculated.
Models
InstaNovo 1.0.0 includes a new model instanovo_extended.ckpt trained on a larger dataset with more
PTMs
Training Datasets
- ProteomeTools Part
I (PXD004732),
II (PXD010595), and
III (PXD021013) \
(referred to as the all-confidence ProteomeTools
AC-PTdataset in our paper) - Additional PRIDE dataset with more modifications: \ (PXD000666, PXD000867, PXD001839, PXD003155, PXD004364, PXD004612, PXD005230, PXD006692, PXD011360, PXD011536, PXD013543, PXD015928, PXD016793, PXD017671, PXD019431, PXD019852, PXD026910, PXD027772)
- Additional phosphorylation dataset \ (not yet publicly released)
Natively Supported Modifications
- Oxidation of methionine
- Cysteine alkylation / Carboxyamidomethylation
- Asparagine and glutamine deamidation
- Serine, Threonine, and Tyrosine phosphorylation
- N-terminal ammonia loss
- N-terminal carbamylation
- N-terminal acetylation
See residue configuration under instanovo/configs/residues/extended.yaml
Additional features
Spectrum Data Class
InstaNovo introduces a Spectrum Data Class: SpectrumDataFrame.
This class acts as an interface between many common formats used for storing mass spectrometry,
including .mgf, .mzml, .mzxml, and .csv. This class also supports reading directly from
HuggingFace, Pandas, and Polars.
When using InstaNovo, these formats are natively supported and automatically converted to the
internal SpectrumDataFrame supported by InstaNovo for training and inference. Any data path may be
specified using glob notation. For example you
could use the following command to get de novo predictions from all the files in the folder
./experiment:
python -m instanovo.transformer.predict data_path=./experiment/*.mgfAlternatively, a list of files may be specified in the inference config.
The SpectrumDataFrame also allows for loading of much larger datasets in a lazy way. To do this, the
data is loaded and stored as .parquet files in a
temporary directory. Alternatively, the data may be saved permanently natively as .parquet for
optimal loading.
Example usage:
Converting mgf files to the native format:
from instanovo.utils import SpectrumDataFrame
# Convert mgf files native parquet:
sdf = SpectrumDataFrame.load("/path/to/data.mgf", lazy=False, is_annotated=True)
sdf.save("path/to/parquet/folder", partition="train", chunk_size=1e6)Loading the native format in shuffle mode:
# Load a native parquet dataset:
sdf = SpectrumDataFrame.load("path/to/parquet/folder", partition="train", shuffle=True, lazy=True, is_annotated=True)Using the loaded SpectrumDataFrame in a PyTorch DataLoader:
from instanovo.transformer.dataset import SpectrumDataset
from torch.utils.data import DataLoader
ds = SpectrumDataset(sdf)
# Note: Shuffle and workers is handled by the SpectrumDataFrame
dl = DataLoader(
ds,
collate_fn=SpectrumDataset.collate_batch,
shuffle=False,
num_workers=0,
)Some more examples using the SpectrumDataFrame:
sdf = SpectrumDataFrame.load("/path/to/experiment/*.mzml", lazy=True)
# Remove rows with a charge value > 3:
sdf.filter_rows(lambda row: row["precursor_charge"]<=2)
# Sample a subset of the data:
sdf.sample_subset(fraction=0.5, seed=42)
# Convert to pandas
df = sdf.to_pandas() # Returns a pd.DataFrame
# Convert to polars LazyFrame
lazy_df = sdf.to_polars(return_lazy=True) # Returns a pl.LazyFrame
# Save as an `.mgf` file
sdf.write_mgf("path/to/output.mgf")SpectrumDataFrame Features:
- The SpectrumDataFrame supports lazy loading with asynchronous prefetching, mitigating wait times between files.
- Filtering and sampling may be performed non-destructively through on file loading
- A two-fold shuffling strategy is introduced to optimise sampling during training (shuffling files and shuffling within files).
Using your own datasets
To use your own datasets, you simply need to tabulate your data in either Pandas or Polars with the following schema:
The dataset is tabular, where each row corresponds to a labelled MS2 spectra.
sequence (string)\ The target peptide sequence including post-translational modificationsmodified_sequence (string) [legacy]\ The target peptide sequence including post-translational modificationsprecursor_mz (float64)\ The mass-to-charge of the precursor (from MS1)charge (int64)\ The charge of the precursor (from MS1)mz_array (list[float64])\ The mass-to-charge values of the MS2 spectrumintensity_array (list[float32])\ The intensity values of the MS2 spectrum
For example, the DataFrame for the nine species benchmark dataset (introduced in Tran et al. 2017) looks as follows:
| sequence | modified_sequence | precursor_mz | precursor_charge | mz_array | intensity_array | |
|---|---|---|---|---|---|---|
| 0 | GRVEGMEAR | GRVEGMEAR | 335.502 | 3 | [102.05527 104.052956 113.07079 ...] | [ 767.38837 2324.8787 598.8512 ...] |
| 1 | IGEYK | IGEYK | 305.165 | 2 | [107.07023 110.071236 111.11693 ...] | [ 1055.4957 2251.3171 35508.96 ...] |
| 2 | GVSREEIQR | GVSREEIQR | 358.528 | 3 | [103.039444 109.59844 112.08704 ...] | [801.19995 460.65268 808.3431 ...] |
| 3 | SSYHADEQVNEASK | SSYHADEQVNEASK | 522.234 | 3 | [101.07095 102.0552 110.07163 ...] | [ 989.45154 2332.653 1170.6191 ...] |
| 4 | DTFNTSSTSN(+.98)STSSSSSNSK | DTFNTSSTSN(+.98)STSSSSSNSK | 676.282 | 3 | [119.82458 120.08073 120.2038 ...] | [ 487.86942 4806.1377 516.8846 ...] |
For de novo prediction, the sequence column is not required.
We also provide a conversion script for converting to native SpectrumDataFrame (sdf) format:
usage: python -m instanovo.utils.convert_to_sdf source target [-h] [--is_annotated IS_ANNOTATED] [--name NAME] [--partition {train,valid,test}] [--shard_size SHARD_SIZE] [--max_charge MAX_CHARGE]
positional arguments:
source source file(s)
target target folder to save data shards
options:
-h, --help show this help message and exit
--is_annotated IS_ANNOTATED
whether dataset is annotated
--name NAME name of saved dataset
--partition {train,valid,test}
partition of saved dataset
--shard_size SHARD_SIZE
length of saved data shards
--max_charge MAX_CHARGE
maximum charge to filter outNote: the target path should be a folder.
ToDo:
- Multi-GPU support
License
Code is licensed under the Apache License, Version 2.0 (see LICENSE)
The model checkpoints are licensed under Creative Commons Non-Commercial (CC BY-NC-SA 4.0)
BibTeX entry and citation info
@article{eloff_kalogeropoulos_2024_instanovo,
title = {De novo peptide sequencing with InstaNovo: Accurate, database-free peptide identification for large scale proteomics experiments},
author = {Kevin Eloff and Konstantinos Kalogeropoulos and Oliver Morell and Amandla Mabona and Jakob Berg Jespersen and Wesley Williams and Sam van Beljouw and Marcin Skwark and Andreas Hougaard Laustsen and Stan J. J. Brouns and Anne Ljungars and Erwin Marten Schoof and Jeroen Van Goey and Ulrich auf dem Keller and Karim Beguir and Nicolas Lopez Carranza and Timothy Patrick Jenkins},
year = {2024},
doi = {10.1101/2023.08.30.555055},
publisher = {Cold Spring Harbor Laboratory},
URL = {https://www.biorxiv.org/content/10.1101/2023.08.30.555055v3},
journal = {bioRxiv}
}Acknowledgements
Big thanks to Pathmanaban Ramasamy, Tine Claeys, and Lennart Martens for providing us with additional phosphorylation training data.