LaMIRA
Lariat Mapping by Inverted Read Alignment
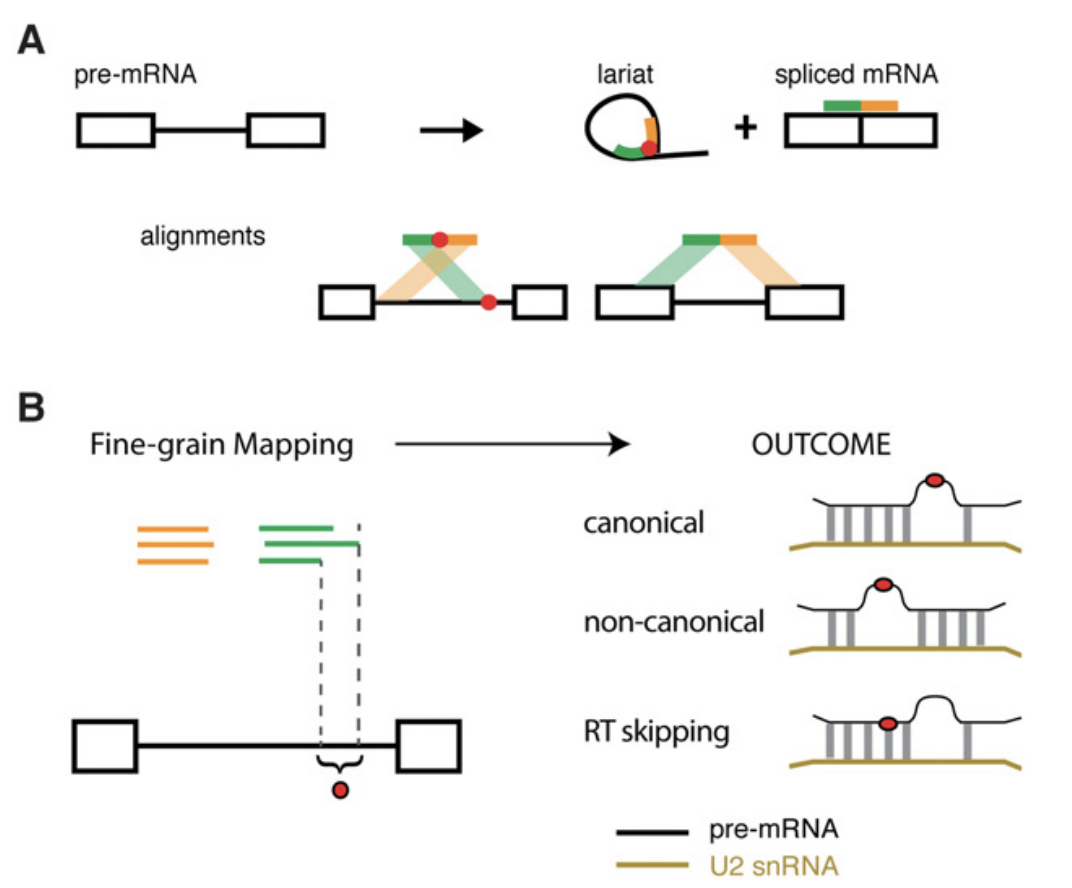
This pipeline identifies RNA-seq reads that originate from lariats. Through iterative alignment, reads are called that contain adjacent segments that map to a 5' splice site and a downstream intronic segment (see figure below). This inverted alignment is a result of RT transcribing from a lariat and reading through the branchpoint. Once lariat reads are identified, post-processing scripts analyze the branchpoints implied by the read mapping.
This pipeline was developed by Allison Taggart and Luke Buerer. It implements the algorithm described in the Supplemental Methods of 'Large-scale analysis of branchpoint usage across species and cell lines'.
Dependencies
Requires the following:
- perl (tested with 5.20.2)
- python3 (tested with 3.7.6)
- bowtie (tested with 1.1.1)
- bedtools (tested with 2.29.2)
-
python-Levenshtein (tested with 0.12.0)
Usage
The pipeline is split into two steps. First, lariat mapping is performed by
map_lariats.sh. This is the computationally expensive step so dedicate more threads to it for faster processing.bash map_lariats.sh <read_file> <read_len> <bowtie_index> <gtf_file> <threads> <out_dir>Option Description read_file Single fastq file that has been QC and adaptor-trimmed to a uniform length (paired-end files should be processed separately) read_len Length of the reads in read_file bowtie_index Basename of bowtie-indexed genome (i.e. /path/to/dir/genome where /path/to/dir/genome.fa is the genome fasta) gtf_file GTF file containing annotated exons threads The number of threads available for use out_dir Writes output to <out_dir>/lariat_reads\ The next step uses
process_BPs.shto analyze the lariat reads identified bymap_lariats.shand output the final branchpoint table. This step requires fewer resources so it should be fine with just one or two threads.bash process_BPs.sh <bowtie_index> <out_dir> <read_len> <threads>Option Description bowtie_index Basename of bowtie-indexed genome (i.e. /path/to/dir/genome where /path/to/dir/genome.fa is the genome fasta) out_dir Writes output to <out_dir>/bp_processingread_len Length of reads threads The number of threads available for use
Output
Lariat read mapping data is output to <out_dir>/lariat_reads/lariat_data_table.txt.
| Column | Description |
|---|---|
| 1 | Sample lariat is from |
| 2 | Inverted alignment type |
| 3 | Read ID |
| 4 | Raw read sequence |
| 5 | Chromosome |
| 6 | Strand |
| 7 | 5' splice site coordinate |
| 8 | 3' splice site coordinate |
| 9 | Branchpoint coordinate |
| 10 | Raw branchpoint sequence (10 nt window, position 5 is the BP) |
\
The final analyzed branchpoint data is output to <out_dir>/bp_processing/BP_final_table.txt.
| Column | Name | Description |
|---|---|---|
| 1 | chrom | Chromosome of the branchpoint |
| 2 | coord | Coordinate of the branchpoint |
| 3 | strand | Strand of the branchpoint |
| 4 | model | Model of branchpoint motif, one of: canonical, canonical2nt, canonicalC, TRAYTRY, TRANYTRY, none, circle, or template_switching |
| 5 | bp_seq | Branchpoint Sequence - parentheses around bulge, BP nucleotide is left of * |
| 6 | bp_nt | Branchpoint nucleotide |
| 7 | threep_ss | Closest 3' splice site downstream of branchpoint coordinate |
| 8 | threep_dist | Distance from branchpoint to 3' splice site |
| 9 | bp_pos | Branchpoint distance category, one of: proximal (BP is between -1 and -10bps upstream of 3'SS), expected (BP is between -11 and -60bps upstream of 3'SS), distal (BP is >60bps upstream of 3'SS), circle (BP is an annotated 3'SS) |
| 10 | total_reads | The total number of reads supporting this branchpoint |
| 11 | unique_reads | The number of unique reads supporting this branchpoint |
| 12 | mut_qc | Branchpoint mutation present in at least 1 read |
| 13 | multi_qc | Branchpoint discovered in multiple RNA-seq sources |
| 14 | total_reads_pos | Total read count (by position) |
| 15 | unique_reads_pos | Unique Read Count (by position) |
| 16 | total_mut_pos | Read Count with Mutation (by position) |
| 17 | unique_mut_pos | Unique Read Count with Mutation (by position) |
| 18 | sources | RNA-seq experiments with lariat reads for this branchpoint |