![]()
sraX
The proposed tool constitutes a Perl package, composed of functional modules, that allows performing a one-step accurate resistome analysis of assembled sequence data from FASTA files.
![]()
Content
- Introduction
- Installation
- Usage --> Follow this comprehensive Tutorial
- License
- Feedback/Issues
- Citation
Introduction
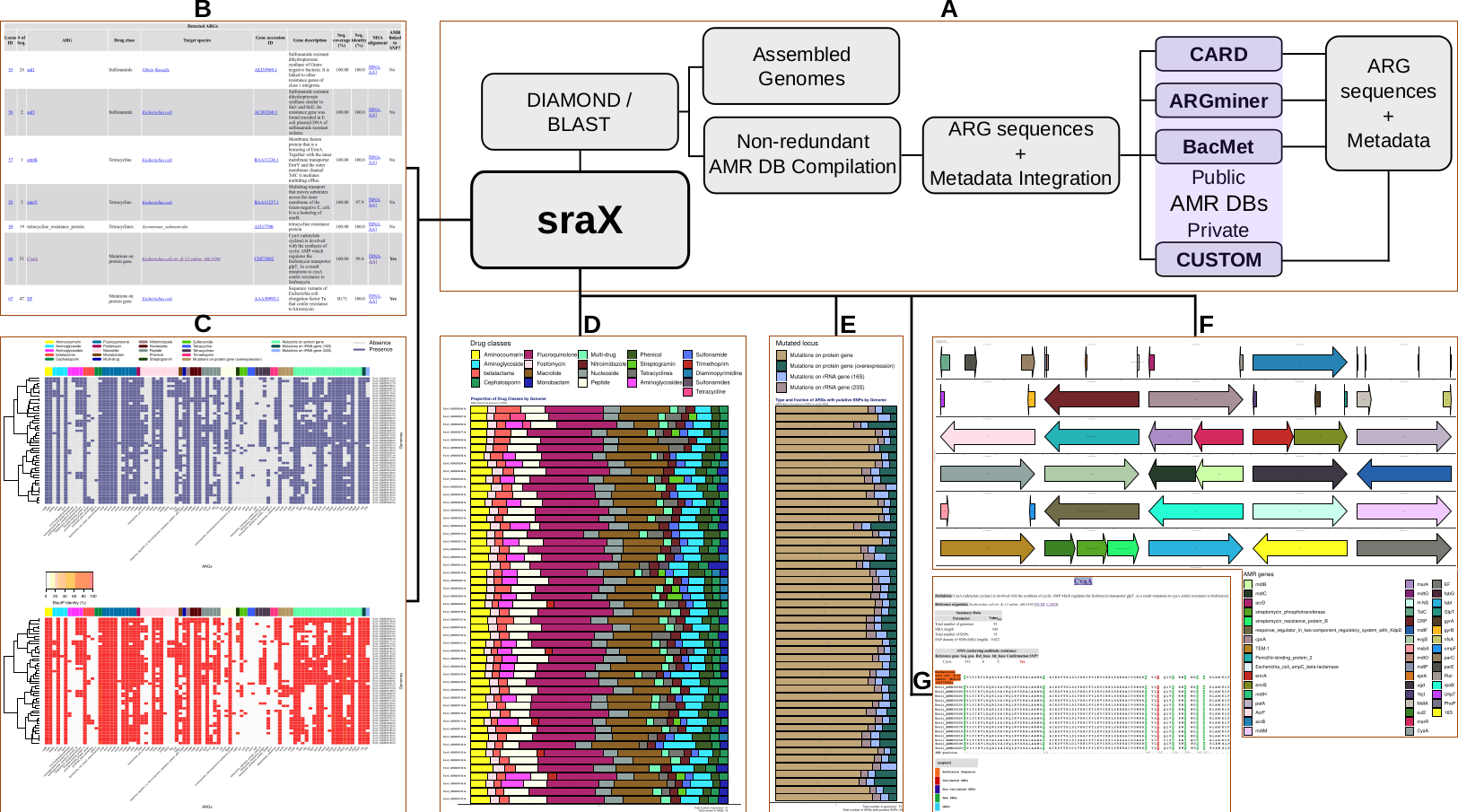
sraX is designed to read assembled sequence files in FASTA format and systematically detect the presence of AMR determinants and, ultimately, describe the repertoire of antibiotic resistance genes (ARGs) within a collection of genomes (the “resistome” analysis). The following assignments are fully automated:
- creation and compilation of a local AMR database (DB) using public or proprietary repositories
- accurate identification of AMR determinants (ARGs or SNPs presence) in a non-redundant manner
- detection of putative new variants through the SNP analysis
- calculation and graphical representation of drug classes and type of mutated loci
- in-depth gene context exploration
The results are presented in fully navigable HTML-formatted files with embedded plots of previously mentioned analysis.
Workflow schematic:
Installation
A) Bioconda / Conda package package:
Execute the following command:
conda install sraxor
conda install -c lgpdevtools sraxVerify the appropriate installation by running:
sraX -vB) Docker image:
Execute the following command:
docker pull lgpdevtools/sraxIn order to check the appropriate running state of the image file:
sudo docker run -it lgpdevtools/srax -vC) Local installation:
sraX has the following dependencies:
Dependencies
1. Though sraX is fully written in Perl and should work with any OS, it has only been tested with a 64-bit Linux distribution.
2. Perl version 5.26.x or higher. You can verify on your own computer by typing the following command in a bash terminal:
perl -hThe latest version of Perl can be obtained from the official website. Consult the installation guide.
-
The following Perl libraries are also required and can be installed using CPAN:
- LWP::Simple
- Data::Dumper
- JSON
- File::Slurp
- FindBin
- Cwd
3. Third-party software
- BLAST (v2.9.0) [1]
- DIAMOND (v0.9.29) [2]
- MUSCLE [3]
- MAFFT (v7.450) [4]
- CLUSTAL Ω (v1.2.4) [5]
- R (v.3.6.1) [6], plus the following packages:
dplyr[7]ggplot2[8]gridExtra[9]
NOTE: The bash script 'install_srax.sh' is provided, in order to confirm
the existence of these dependencies in your computer. If any of them would be
missing, the bash script will guide you for a proper installation.
To successfully install sraX, please see the details provided below. If you encounter an issue during the process, please contact your local system administrator. If you encounter a bug please log it here or email me at lgpanunzi@gmail.com
Open a bash terminal and clone the repository:
git clone https://github.com/lgpdevtools/sraX.gitTo verify the existence of required dependencies and ultimately install the perl modules composing sraX, inside the cloned repository run:
sudo bash install_srax.shUsage:
sraX effectively operates as one-step application. It means that just a single command is required to obtain the totality of results and their visualization.
NOTE: For a detailed explanation and examples from real datasets, please follow the Tutorial.
Parameters
Usage:
-i|input <Mandatory: input genome directory>
-o|output <Optional: name of output folder>
-db|dbsearch <Optional: the level of the ARG search, based on the employed reference AMR DBs (default: basic)>
-s|seqal <Optional: algorithm for aligning the query genome to the reference AMR DB (default: dblastx)>
-a|msa <Optional: algorithm for producing the MSA files (default: muscle)>
-e|eval <Optional: evalue cut-off to filter false positives (default: 1e-05)>
-c|aln_cov <Optional: fraction of aligned query to the reference sequence (default: 60)>
-id <Optional: sequence identity percentage cut-off to filter false positives (default: 85)>
-u|user_sq <Optional: input private AMR DB>
-t|threads <Optional: number of threads to use (default: 6)>
-v|version <print current version>
-d|debug <Optional: print verbose output for debugging (default: No)>
-h|help <print this message>Minimal command
Example usage:
sraX -i [/path/to/input_genome_directory]Where:
-i Full path to the mandatory directory containing the input sequence data, which must
be in FASTA format and consisting of individual assembled genome sequences.Extended options
Example usage:
sraX -a mafft -db ext -s blastx -id 95 -c 90 -t 12 -o [/path/to/output_results_directory] -i [/path/to/input_genome_directory]Docker-based:
sudo docker run --rm -v $(pwd)/[/path/to/input_genome_directory]:/IN lgpdevtools/srax -i INWith further options:
sudo docker run --rm -v $(pwd)/[/path/to/input_genome_directory]:/IN \
-v $(pwd)/[/path/to/output_results_directory]:/OUT \
lgpdevtools/srax -a mafft -db ext -s blastx -id 95 -c 90 -t 12 -i IN -o OUTWhere:
Mandatory:
----------
-i|input Input directory [/path/to/input_dir] containing the input file(s), which
must be in FASTA format and consisting of individual assembled genome sequences.
Optional:
---------
-o|output Directory to store obtained results [/path/to/output_dir]. While not
provided, the following default name will be taken:
'input_directory'_'sraX'_'id'_'aln_cov'_'seqal'
Example:
--------
Input directory: 'Test'
Options: -id 85; -c 95; -p dblastx
Output directory: 'Test_sraX_85_95_dblastx'
-s|seqal The preferred algorithm for aligning the assembled genome(s) to a locally
compiled AMR DB. The possible choices are: 'dblastx' (DIAMOND blastx) or 'blastx'
(NCBI blastx). In any case, the process is parallelized (up to 100 genome files are
run simultaneously) for reducing computing times. [string] Default: dblastx
-a|msa The preferred algorithm for producing the alignment of clustered homologous
sequences (multiple-sequence files). The possible choices are: 'muscle', 'clustalo'
or 'mafft'. [string] Default: muscle
Note: The accuracy and computing times are both dependent on the selected algorithm.
-e|eval Minimum evalue cut-off to filter false positives. [number] Default: 1e-05
-id Minimum identity cut-off to filter false positives. [number] Default: 85
-c|aln_cov Minimum length of the query which must align to the reference sequence.
[number] Default: 60
-db|dbsearch The level of the ARG search, on account of the number and type of employed AMR DB.
The possible choices are: 'basic' or 'ext' / 'extensive'. The
'basic' option only applies 'CARD', while the 'ext' option utilizes as well the
'ARGminer' (compilation of multiple AMR DBs) and 'BACMET'
(biocides and metal resistance) repositories. [string] Default: basic
Note: In operational terms, the extensive search ('ext' option) takes much longer
computing times.
-u|user_sq Customary AMR DB provided by the user. The sequences must be in FASTA format.
-t|threads Number of threads when running sraX. [number] Default: 6
-h|help Displays this help information and exits.
-v|version Displays version information and exits.
-d|debug Verbose output (for debugging).
License
sraX is free software, licensed under GPLv3.
Feedback/Issues
Please report any issues to the issues page or email lgpanunzi@gmail.com
About
sraX is developed by Leonardo G. Panunzi.
Citation
Panunzi LG, sraX: a novel comprehensive resistome analysis tool, submitted to Frontiers in Microbiology for publication.
References.
[1] Altschul SF et al. (1990). Basic local alignment search tool. JMB, 215, 403–410.
[2] Buchfink B, Xie C & Huson DH (2015). Fast and sensitive protein alignment using DIAMOND. Nature Methods 12, 59-60.
[3] Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5):1792-1797.
[4] Katoh et al. (2002). Mafft: a novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic acids research 30, 3059–3066.
[5] Sievers F. et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Molecular systems biology 7, 539.
[6] R Core Team (2013). R: A Language and Environment for Statistical Computing.
[7] Wickham H, Romain Francois R, Henry L and Müller K (2017). dplyr: A Grammar of Data Manipulation.
[8] Wickham H (2016). ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York.
[9] Auguie B, Antonov A and Auguie MB (2016).