
Easy353: An Efficient Tool Designed to Recover the Angiosperms353 Gene Set
Notice: The Easy353 has been updated to v2.0.1, which is faster and more accurate.
NOTE: We have specifically developed a enhanced graphical interface version, EasyMiner, for Windows and macOS (developing). Please visit EasyMiner on GitHub or Gitee for details.
Please cite the following manuscript if you use the Easy353:
Zhen Zhang, Pulin Xie, Yongling Guo, Wenbin Zhou, Enyan Liu, Yan Yu. Easy353: A tool to get Angiosperms353 genes for phylogenomic research. Molecular Biology and Evolution. msac261 (2022). https://doi.org/10.1093/molbev/msac261.
Additionally, please cite the dependencies if used:
Baker W.J., Bailey P., Barber V., Barker A., Bellot S., Bishop D., Botigue L.R., Brewer G., Carruthers T., Clarkson J.J., Cook J., Cowan R.S., Dodsworth S., Epitawalage N., Francoso E., Gallego B., Johnson M., Kim J.T., Leempoel K., Maurin O., McGinnie C., Pokorny L., Roy S., Stone M., Toledo E., Wickett N.J., Zuntini A.R., Eiserhardt W.L., Kersey P.J., Leitch I.J. & Forest F. A Comprehensive Phylogenomic Platform for Exploring the Angiosperm Tree of Life. Systematic Biology. 71: 301–319. https://doi.org/10.1093/sysbio/syab035.
Easy353 is a tool specifically designed to recover the Angiosperms353 gene set (AGS). It effectively filters AGS-related reads from high-throughput sequencing data, and accurately recovers AGS using its optimized reference-guided assembler.
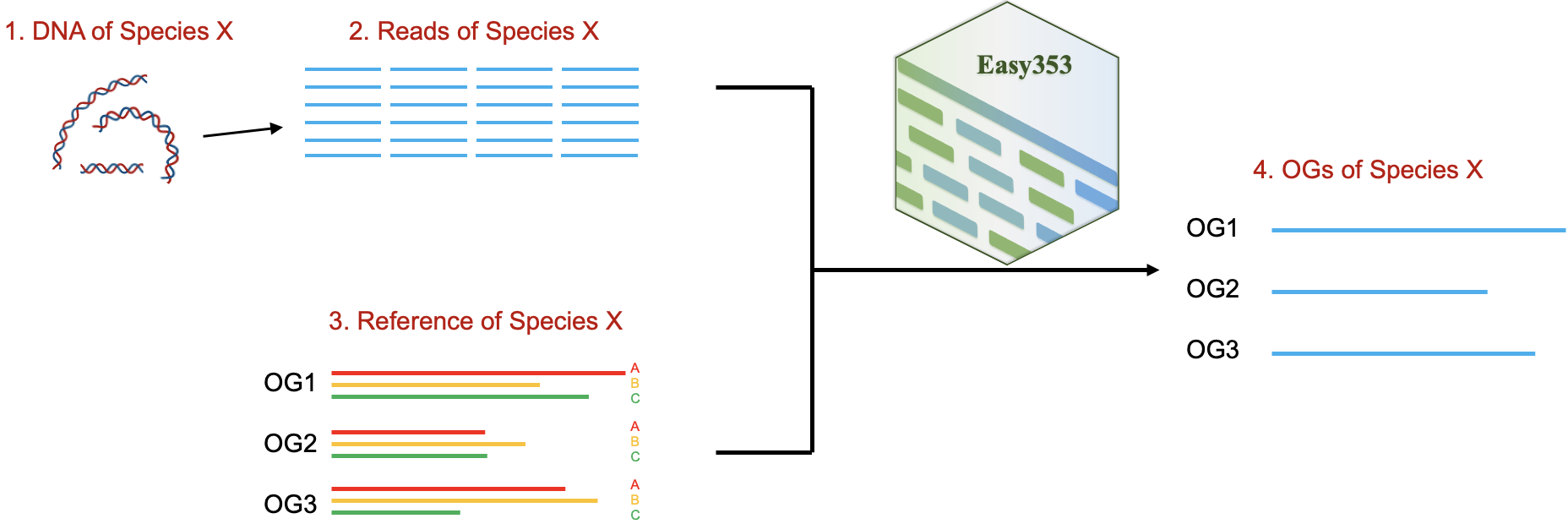
To run Easy353, two things are required as input: the sequencing data (reads in FASTQ format) and a set of reference sequences (orthologous genes from other species). The target genes of species X are included in the output file. Notice: Easy353 can recover not only AGS but also user-specified target genes (e.g., chloroplast genes, ITS). However, if the target gene is a gene like ITS, the user must provide their own reference sequences. Using the script build_database.py, users can download the AGS reference sequences while recovering AGS.
Download and Installation
Easy353 is a user-friendly tool developed in Python 3 (3.6 and above), offering two interfaces: a full graphical user interface (Easy353-GUI) and a command-line interface (Easy353).
Easy353-GUI
For Windows and macOS users, it is recommended to download the ALL-IN-ONE graphical user interface version of Easy353 (Easy353-GUI) from Easy353/releases.
Easy353-cmd
There are several generally 2 ways to install Easy353-cmd:
- Option 1 Using the setup.py
- Option 2 In situ configuration
Option 1. Using the setup.py
The most up-to-date version of Easy353 is available at our GitHub site plant720/Easy353. Users should use git to download the entire Easy353 repository and install the Easy353 using the setup.py.
- Clone the Easy353 repository:
# get a local copy of the easy353 source code
git clone https://github.com/plant720/Easy353.git- Install the code:
# install the code
cd Easy353
python3 setup.py install --user- For some Linux and macOS systems, after executing the above commands, you may not be able to run
build_database.pyandeasy353.pydirectly in a new terminal, indicating that~/.local/binhas not been added to the$PATH. In this case, you have to manually add~/.local/bin:
# add ~/.local/bin to PATH
echo "PATH=~/.local/bin:\$PATH" >> ~/.bashrc
source /.bashrcOption 2. In situ configuration
Alternatively, you can use the following commands to download and configure Easy353 in situ:
- Clone the Easy353 repository and create a directory for installation:
# Assuming you want to install it at ~/Applications
mkdir ~/Applications # create directories if not exist
cd ~/Applications
git clone https://github.com/plant720/Easy353.git- Make the Easy353 scripts executable:
chmod +x Easy353/build_database.py
chmod +X Easy353/easy353.py- Add Easy353 to the
$PATH.
echo "export PATH=~/Applications/Easy353:\$PATH" >> ~/.bashrc
source ~/.bashrc- Install the required Python libraries, including biopython, psutil, requests, and beautifulsoup4:
# install required libs
pip install biopython psutil requests beautifulsoup4Update
This section provides a step-by-step guide on how to update Easy353. The update process involves two main steps: uninstalling the old version and installing the new one.
1. Uninstallation
The method for uninstallation depends on how Easy353 was initially installed.
Option 1: Using the setup.py
-
Uninstall easy353 using pip3:
- Run the command
pip3 uninstall easy353to remove the installed package.
- Run the command
-
Delete the Downloaded Folder:
- Remove the previously downloaded Easy353 folder from your system.
Option 2: In situ Configuration
- Direct Deletion:
- Simply delete the previously downloaded Easy353 folder from your system.
2. Installation of the New Version
After successfully uninstalling the old version:
-
Download the Latest Version:
- Use Git to clone or download the new version of Easy353.
-
Follow Installation Steps:
- Proceed with the installation steps as described in the section of
Download and Installation.
- Proceed with the installation steps as described in the section of
Usage
Note: The detailed usage and tutorials can be found at Easy353/wiki.
After installation, Easy353 can be used to filter and assemble reads to recover target genes.
- Prepare the input:
To run Easy353, two things are required as the input: the sequencing reads in FASTQ format and a set of reference sequences, i.e. AGS. The quality of the reference sequences strongly influences the Easy353's result. Hence, it is crucial to generate the reference sequences carefully. If the target sequence is AGS, users can use the build_database.py script to generate. However, if the target gene is other marker genes, the users need to generate the reference sequences independently. Here is an example of how to use our script to generate the reference:
# To ensure that the downloaded AGS data consists of sequences from closely related species, please download the sequences according to taxonomy.
# The reference sequences are downloaded from Kew Tree of Life Explorer (https://treeoflife.kew.org), so ensure your device is connected to the network.
build_database.py -o 353_ref_Fabaceae -c Fabaceae -t 10 -exclude Glycine_max -generate
# The final reference sequences can be found in the 353_ref_Fabaceae/353gene directory after downloading
## Explanation of parameters
-o: specifies the output directory
-c: specifiles the taxonomy of species used as reference
-t: specifies the number of threads used to download files
-exclude: excludes species that are not used as reference
-generate: generates a CSV that records the information about the downloaded species- Run the Easy353, the following is an example of the command syntax:
easy353.py -1 <input_file1> -2 <input_file2> -r <reference_dir> -o <output_dir> -fk <filter_kmer> -ak <assemble_kmer> -ft <filter_thread> -at <assemble_thread>
## The parameters used in the command are explained below:
-1 -2: the input files with paired-end reads, given in FASTQ format.
-r: the reference directory
-o: the output directory
-fk: K-mer length setting for filtering
-ak: K-mer length setting for assembly
-ft: the threads setting for reads filtering
-at: the threads setting for reads assembly
# An example
easy353.py -1 Gmax_sim_1.fastq.gz -2 Gmax_sim_2.fastq.gz -r 353_ref_Fabaceae/353gene -o test_package -fk 31 -ak 41 -ft 1 -at 4- Now, you can view the result of Easy353 within the output directory:
The output directory comprises two subdirectories: filtered_reads and target_genes.
- The
filtered_outsubdirectory contains filtered reads associated with target genes. - The
assemble_outsubdirectory is the primary result directory of Easy353, storing target gene sequences recovered by the tool.
Easy353-GUI
Note: There is a detailed guide of Easy353-GUI at
Please use the Easy353-gui.app or Easy353-gui.exe from https://github.com/plant720/Easy353/releases, the Easy353-gui.py is only used for development.
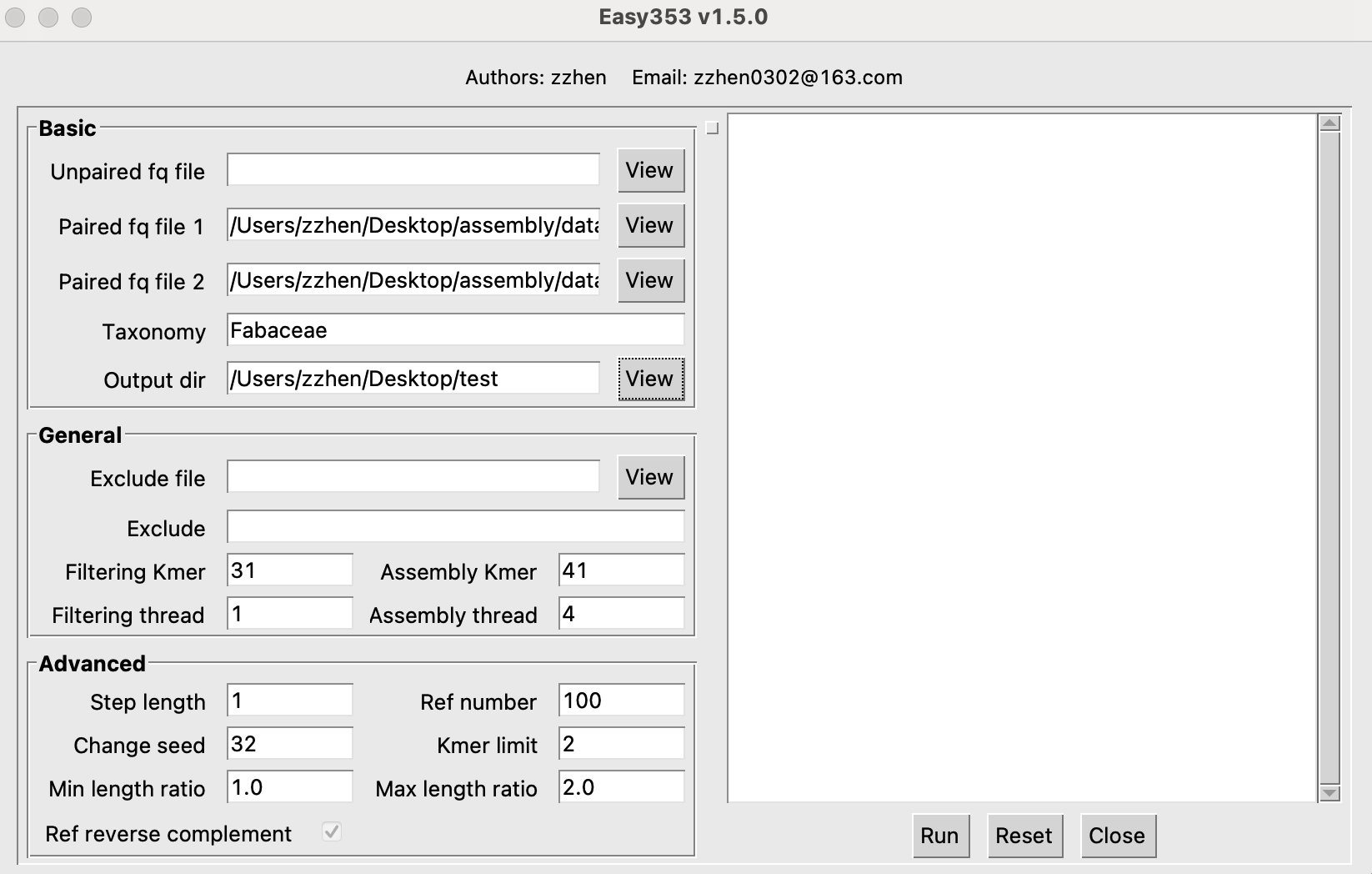
- For Easy353-GUI, you should set the
Paired fq file 1,Paired fq file 2,Taxonomy,andOutput diras shown in the image below
Contact
If you have any questions, suggestions, or comments about Easy353, feel free to contact the developer at zzhen0302@163.com.