scCancer
Introduction
The scCancer package focuses on processing and analyzing droplet-based scRNA-seq data for cancer research. Except basic data processing steps, this package takes several special considerations for cancer-specific features.
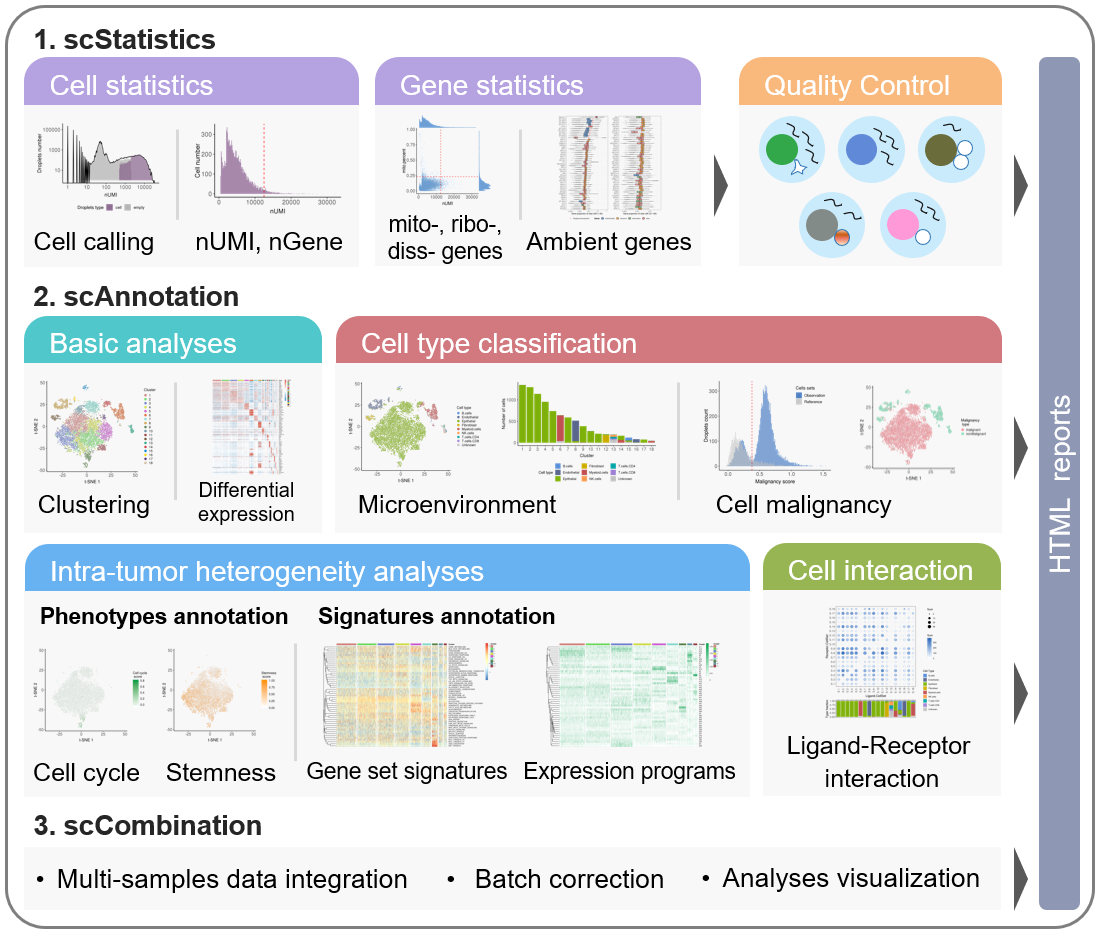
The workflow of scCancer mainly consists of three modules: scStatistics, scAnnotation, and scCombination.
- The
scStatisticsperforms basic statistical analyses of raw data and quality control. - The
scAnnotationperforms functional data analyses and visualizations, such as low dimensional representation, clustering, cell type classification, cell malignancy estimation, cellular phenotype analyses, gene signature analyses, cell-cell interaction analyses, etc. - The
scCombinationperform multiple samples data integration, batch effect correction and analyses visualization.
After the computational analyses, detailed and graphical reports were generated in user-friendly HTML format.
(Click to view larger workflow picture)
System Requirements
- R version: >= 3.5.0 (suggest: R 3.6, not 4.0)
- Hint: For R (version>=4.0) under Windows system, the Rtools needs to be updated to version 4.0 from https://cran.r-project.org/bin/windows/Rtools/. So, if you are not familiar with R environment configuration, we don't suggest to use R (>=4.0).
Current version
- scCancer 2.2.1 (update at 2021.03.02)
- All version log
Installation
The detailed installation instruction can be found in the project wiki.
Usage
The vignette of scCancer can be found in the project wiki.
We provide an example data of kidney cancer from 10X Genomics, and following are the generated HTML reports:
For multi-datasets, following is a generated HTML report for three kidney cancer samples integration analysis:
Citation
Please use the following citation:
[1] Wenbo Guo, Dongfang Wang, Shicheng Wang, Yiran Shan, Changyi Liu, Jin Gu, scCancer: a package for automated processing of single-cell RNA-seq data in cancer, Briefings in Bioinformatics, bbaa127, https://doi.org/10.1093/bib/bbaa127
[2] Zeyu Chen, Yuxin Miao, Zhiyuan Tan, Qifan Hu, Yanhong Wu, Xinqi Li, Wenbo Guo, Jin Gu, scCancer2: data-driven in-depth annotations of the tumor microenvironment at single-level resolution, Bioinformatics, Volume 40, Issue 2, February 2024, btae028, https://doi.org/10.1093/bioinformatics/btae028
License
GPL-3