-
#8, remember EXACTLY P threads ( +- main, whatever) exist.
Currently:
https://github.com/malimban/dna/blob/e6d90a4cae9d79af873abfdab46f691afd439c16/par.java#L64-L78
https://github.com/malimb…
-
Can I remove the display of some DNA in metadata using these lines of code, if so, how?
(I'm not a programmer)
(The code is located in main.js v 1.1.2)
[](url)
:
File "/genetics-master/hello_world.py", line 42, in
best, best_score, average_score = dna_…
unnir updated
7 years ago
-
https://doi.org/10.1101/172767 (http://www.biorxiv.org/content/early/2017/08/06/172767)
> Determining the binding locations of regulatory factors, such as transcription factors and histone modifica…
-
Latest GPML for [pathwayPA165292163](https://www.pharmgkb.org/pathway/PA165292163) is obsolete.
Use this instead: [PA165292163-new.gpml](https://drive.google.com/a/pharmgkb.org/uc?export=download&id=1…
-
Hi there!
I have a problem calculating the major/minor groove distances of my DNA. My DNA is 70 bp long, but I do get as amount of major grooves around 64, which is way too much compared to my DNA s…
-
Apparently the information can be read from SnapGene `.dna` files as xml. This would be important to do, since it will allow people familiar with SnapGenes' UI to do the cloning there, but still share…
-
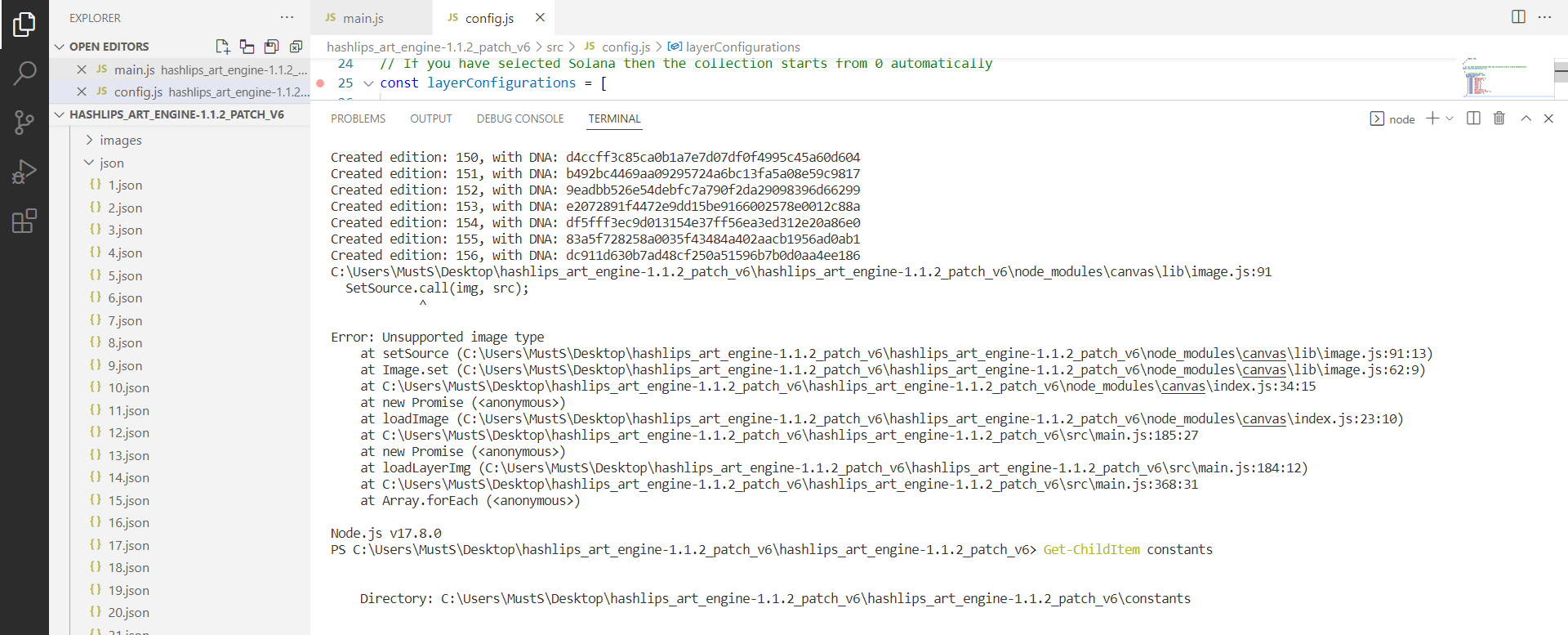
Everything was running fine than this started happening
Created edition: 151, w…