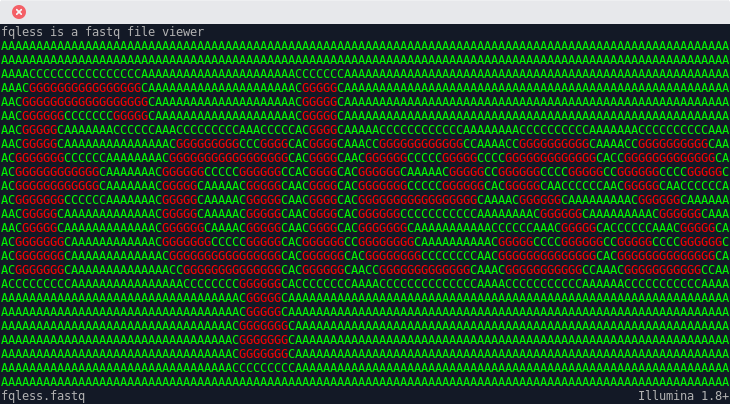
fqless - FastQ Less
fqless is intended to be a small less-like viewer for FastQ sequencing files. Allowing the user to get a quick glance at their sequencing data without the need of a heavy GUI/Java-bloatware.
It displays the name and the color coded sequence. It hides the quality line from the user, as this is machine information for machines and not for human beings.
It has no problem opening many gigabytes of fastq files, as it will not load everything, but will be buffering based on an index, it builds in memory. Thus after opening a large file, you will see high CPU usage until the index is build, but you can already see everything.
fqless is released under the GPLv2 or any newer version of the GPL. It comes with no warranty. Use it as you wish.
Use it via
fqless file.tar.gzSee all options via -h flag.
How to build
On UNIX:
git clone https://github.com/openpaul/fqless
cd fqless
./configure
make
make installActivate color support in your terminal
To color code the DNA fqless uses ncurses which requires you to use a 256 color compatible terminal. Most modern terminal emulators have this build in but sometimes this needs to be activated in your .bashrc by inserting the line TERM=xterm-256color.
On Windows:
Install UNIX then GOTO UNIX.
Seriously: I don't have access to a Windows PC and no interest in porting this to windows. If you want to make a pull request with a working patch, feel free to do so.
What it does
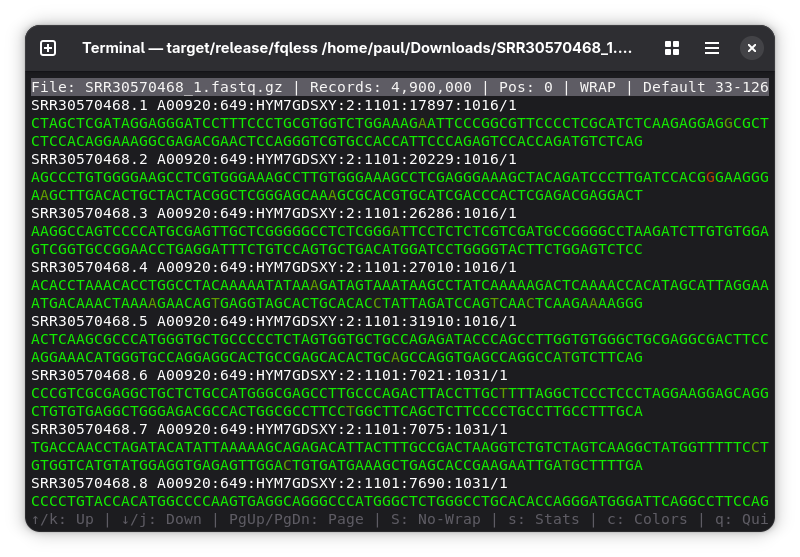
- Opens the file, shows the sequence color coded, so one can decide if the run quality is nice and fits the expectations. It tries to guess the quality encoding of the file but you can also change that live, if it guessed the wrong one.
- Move by arrow keys. Change quality encoding by right and left arrow.
What it does not
It does not allow to do anything else (no mapping, no trimming, no whatsoever).
KISS: Keep it simple, stupid.
Features
- Parse FastQ [done]
- read plain text as well as tar.gz [done]
- Detect encoding [done]
- Color code sequence [done]
- allow switching between encodings, only if multiple encodings are in question [done]
- allows conversion to other encodings [maybe]
- allow STDIN [canceled, because, who wants that?]
- write fastq, allow conversion between encodings [todo, maybe]
- write fasta [todo, maybe]
Bugs
This software has most certainly some heavy bugs. I am testing a lot, but will probably not catch everything. So I am very glad if you submit issues. Best would be to supply a minimal working, or not worging, fastq file, that causes the problem.
fqless messes up the colors of the terminal
fqless makes use of ncurses to define needed colors. These are at the moment not reset after fqless terminates and persist in the terminal. So other coloring operations might make use of the colors until the terminal is closed.
Colors do not work or work incorrectly in screen or tmux
Yes, this is unfortunate but apparently not easily solvable at the moment.